
Войти
Технологии транскриптомики - это методы, используемые для изучения транскриптома организма, суммы всех его Транскрипты РНК. Информационное содержание организма записывается в ДНК его генома и , выраженного посредством транскрипции. Здесь мРНК служит временной промежуточной молекулой в информационной сети, в то время как некодирующие РНК выполняют дополнительные разнообразные функции. Транскриптом фиксирует моментальный снимок общего количества транскриптов, присутствующих в ячейке, во времени. Технологии транскриптомики обеспечивают широкое представление о том, какие клеточные процессы активны, а какие бездействуют. Основная проблема молекулярной биологии заключается в понимании того, как один и тот же геном может давать начало различным типам клеток и как регулируется экспрессия генов.
Первые попытки изучить полные транскриптомы начались в начале 1990-х годов. Последующие технологические достижения с конца 1990-х годов неоднократно трансформировали эту область и сделали транскриптомику широко распространенной дисциплиной в биологических науках. В этой области используются два ключевых современных метода: микроматрицы, которые количественно определяют набор заранее определенных последовательностей, и RNA-Seq, в котором используется высокопроизводительное секвенирование для записывать все стенограммы. По мере совершенствования технологии объем данных, производимых каждым экспериментом с транскриптомом, увеличивался. В результате методы анализа данных постоянно адаптируются для более точного и эффективного анализа постоянно растущих объемов данных. Базы данных транскриптомов росли, и их полезность увеличивалась по мере того, как исследователи собирали и распространяли все больше транскриптомов. Было бы почти невозможно интерпретировать информацию, содержащуюся в транскриптоме, без контекста предыдущих экспериментов.
Измерение экспрессии генов организма в различных тканях или условиях, или в разное время, дает информацию о том, как гены регулируется и раскрывает подробности биологии организма. Его также можно использовать для вывода функций ранее неаннотированных генов. Транскриптомный анализ позволил изучить, как экспрессия генов изменяется в различных организмах, и сыграл важную роль в понимании болезни человека. Анализ экспрессии генов в целом позволяет выявить широкие согласованные тенденции, которые нельзя выявить с помощью более целенаправленных анализов.
Для транскриптомики характерно развитие новых методов которые переопределяли то, что возможно, каждые десять лет или около того и сделали предыдущие технологии устаревшими. Первая попытка захвата частичного транскриптома человека была опубликована в 1991 году и сообщила о 609 последовательностях мРНК из человеческого мозга. В 2008 году были опубликованы два человеческих транскриптома, состоящие из миллионов последовательностей, производных от транскриптов, охватывающих 16 000 генов, а к 2015 году были опубликованы транскриптомы для сотен людей. Транскриптомы различных болезненных состояний, тканей или даже отдельных клеток теперь обычно генерируются. Этот взрыв в транскриптомике был вызван быстрым развитием новых технологий с повышенной чувствительностью и экономичностью.
Исследования отдельных транскриптов проводились несколько десятилетий назад. любые подходы к транскриптомике были доступны. Библиотеки транскриптов мРНК шелкопряда были собраны и преобразованы в комплементарную ДНК (кДНК) для хранения с использованием обратной транскриптазы в конце 1970-х годов. В 1980-х годах для секвенирования случайных транскриптов использовалось низкопроизводительное секвенирование с использованием метода Sanger, что дало экспрессируемые теги последовательности (EST). Метод секвенирования по Сэнгеру был преобладающим до появления высокопроизводительных методов, таких как секвенирование путем синтеза (Solexa / Illumina). EST приобрели известность в 1990-х годах как эффективный метод определения содержания гена в организме без секвенирования всего генома. Количества отдельных транскриптов определяли количественно с использованием методов Нозерн-блоттинга, нейлоновых мембранных массивов и более поздних методов количественной ПЦР с обратной транскриптазой (RT-qPCR), но эти методы трудоемки и может захватить только крошечный фрагмент транскриптома. Следовательно, способ, которым транскриптом в целом экспрессируется и регулируется, оставался неизвестным, пока не были разработаны методы с более высокой пропускной способностью.
Слово «транскриптом» впервые было использовано в 1990-х годах. В 1995 году был разработан один из самых ранних транскриптомных методов, основанный на секвенировании, последовательный анализ экспрессии гена (SAGE), который работал посредством секвенирования по Сэнгеру сцепленных случайных фрагментов транскрипта. Транскрипты количественно оценивали путем сопоставления фрагментов с известными генами. Также кратко использовался вариант SAGE с использованием высокопроизводительных методов секвенирования, называемый цифровым анализом экспрессии генов. Однако эти методы в значительной степени уступили место высокопроизводительному секвенированию полных транскриптов, которое предоставило дополнительную информацию о структуре транскриптов, такую как варианты сплайсинга.
| RNA-Seq | Микроматрица | |
|---|---|---|
| Производительность | от 1 дня до 1 недели на эксперимент | 1-2 дня на эксперимент |
| Входное количество РНК | Низкое ~ 1 нг общая РНК | Высокая ~ 1 мкг мРНК |
| Трудоемкость | Высокая (подготовка образцов и анализ данных) | Низкая |
| Предварительные знания | Не требуется, хотя эталонная последовательность генома / транскриптома полезна | Эталонный геном / транскриптом требуется для разработки зондов |
| Количественный анализ точность | ~ 90% ( ограничено охватом последовательностей) | >90% (ограничено точностью обнаружения флуоресценции) |
| Разрешение последовательности | RNA-Seq может обнаруживать SNP и варианты сплайсинга (ограничено точность секвенирования ~ 99%) | Специализированные массивы могут обнаруживать варианты сплайсинга мРНК (ограничены конструкцией зонда и перекрестной гибридизацией) |
| Чувствительность | 1 транскрипт на миллион (приблизительный, ограничен охватом последовательностей) | 1 транскрипт на тысячу (приблизительный, ограничено детектированием флуоресценции) |
| Динамический диапазон | 100000: 1 (ограничено охватом последовательностей) | 1000: 1 (ограничено насыщением флуоресценции) |
| Техническая воспроизводимость | >99% | >99% |
Доминирующие современные методы, микроматрицы и RNA-Seq, были разработаны в середине 1990-х и 2000-х годах. Микроматрицы, которые измеряют численность определенного набора транскриптов посредством их гибридизации с массивом дополнительных зондов, были впервые опубликованы в 1995 году. Технология микроматриц позволяла анализировать тысячи транскриптов одновременно и при значительно меньшей стоимости гена и экономии труда. Оба пятнистых олигонуклеотидных массивов и Affymetrix массивов высокой плотности были методом выбора для транскрипционного профилирования до конца 2000-х годов. В течение этого периода был произведен ряд микроматриц для охвата известных генов модели или экономически важных организмов. Достижения в разработке и производстве наборов улучшили специфичность зондов и позволили тестировать большее количество генов на одном массиве. Достижения в области обнаружения флуоресценции повысили чувствительность и точность измерения для транскриптов с низким содержанием.
RNA-Seq достигается путем обратной транскрипции РНК in vitro и секвенирования полученных кДНК. Обилие транскриптов определяется количеством отсчетов для каждого транскрипта. Таким образом, на эту методику сильно повлияло развитие технологий высокопроизводительного секвенирования. Массивно-параллельное секвенирование сигнатур (MPSS) было ранним примером, основанным на генерации 16–20 п.н. Последовательности посредством сложной серии гибридизаций, которые использовали в 2004 году для проверки экспрессии десяти тысяч генов в Arabidopsis thaliana. Самая ранняя работа по RNA-Seq была опубликована в 2006 году, когда сто тысяч транскриптов секвенировались с использованием технологии 454. Этого покрытия было достаточно для количественной оценки относительного количества транскриптов. Популярность RNA-Seq начала расти после 2008 года, когда новые технологии Solexa / Illumina позволили записать один миллиард последовательностей транскриптов. Этот результат теперь позволяет количественно и сравнивать человеческие транскриптомы.
Получение данных по РНК-транскриптам может быть достигнуто с помощью любого из двух основных принципов: секвенирования отдельные транскрипты (EST или RNA-Seq) или гибридизация транскриптов с упорядоченным массивом нуклеотидных зондов (микроматрицы).
Все транскриптомные методы требуют, чтобы сначала была выделена РНК из экспериментального организма, прежде чем можно будет записать транскрипты. Хотя биологические системы невероятно разнообразны, методы экстракции РНК в целом аналогичны и включают механическое разрушение клеток или тканей, разрушение РНКазы с помощью хаотропных солей, разрушение макромолекул и нуклеотидных комплексов, отделение РНК от нежелательных биомолекул, включая ДНК, и концентрацию РНК посредством осаждения из раствора или элюирования из твердой матрицы. Выделенную РНК можно дополнительно обработать ДНКазой для переваривания любых следов ДНК. Необходимо обогатить матричную РНК, поскольку экстракты общей РНК обычно на 98% состоят из рибосомной РНК. Обогащение транскриптами может быть выполнено методами аффинности поли-A или путем истощения рибосомной РНК с использованием зондов, специфичных для последовательности. Деградированная РНК может повлиять на последующие результаты; например, обогащение мРНК из деградированных образцов приведет к истощению 5 ’концов мРНК и неравномерному сигналу по длине транскрипта. Быстрое замораживание ткани перед выделением РНК является типичным, и после завершения выделения предпринимаются меры для уменьшения воздействия ферментов РНКазы.
328>Метка экспрессированной последовательности (EST) представляет собой короткую нуклеотидную последовательность, полученную из одного транскрипта РНК. РНК сначала копируется как комплементарная ДНК (кДНК) ферментом обратной транскриптазой до секвенирования полученной кДНК. Поскольку EST можно собирать без предварительного знания организма, из которого они происходят, их можно приготовить из смесей организмов или образцов окружающей среды. Хотя сейчас используются высокопроизводительные методы, библиотеки EST обычно предоставляли информацию о последовательностях для ранних дизайнов микрочипов; например, микроматрица ячменя была сконструирована из 350 000 секвенированных ранее EST.
 Краткое изложение SAGE. Внутри организмов гены транскрибируются и сплайсируются (в эукариотах ) для получения зрелых транскриптов мРНК (красный). МРНК извлекается из организма, и обратная транскриптаза используется для копирования мРНК в стабильную двухцепочечную кДНК (ds -кДНК ; синий). В SAGE ds-кДНК расщепляется рестрикционными ферментами (в положениях «X» и «X» + 11) с образованием 11-нуклеотидных «меточных» фрагментов. Эти теги объединяются и секвенируются с использованием длинного считывания секвенирования по Сэнгеру (разные оттенки синего указывают теги из разных генов). Последовательности деконволюционируют, чтобы найти частоту каждого тега. Частота метки может использоваться для сообщения о транскрипции гена, из которого произошла метка.
Краткое изложение SAGE. Внутри организмов гены транскрибируются и сплайсируются (в эукариотах ) для получения зрелых транскриптов мРНК (красный). МРНК извлекается из организма, и обратная транскриптаза используется для копирования мРНК в стабильную двухцепочечную кДНК (ds -кДНК ; синий). В SAGE ds-кДНК расщепляется рестрикционными ферментами (в положениях «X» и «X» + 11) с образованием 11-нуклеотидных «меточных» фрагментов. Эти теги объединяются и секвенируются с использованием длинного считывания секвенирования по Сэнгеру (разные оттенки синего указывают теги из разных генов). Последовательности деконволюционируют, чтобы найти частоту каждого тега. Частота метки может использоваться для сообщения о транскрипции гена, из которого произошла метка. Серийный анализ экспрессии гена (SAGE) был развитием методологии EST для увеличения пропускной способности сгенерированы теги и позволяют некоторое количественное определение количества транскриптов. кДНК генерируется из РНК, но затем расщепляется на «теговые» фрагменты размером 11 п.н. с использованием рестрикционных ферментов, которые разрезать ДНК по определенной последовательности и 11 пар оснований от этой последовательности. Эти теги кДНК затем соединяются в длинные цепи (>500 п.н.) и секвенируются с использованием методов с низкой пропускной способностью, но с большой длиной считывания, таких как секвенирование по Сэнгеру. Затем последовательности делятся обратно на их исходные теги размером 11 пар оснований с использованием компьютерного программного обеспечения в процессе, называемом деконволюцией. Если доступен эталонный геном , эти теги могут быть сопоставлены с их соответствующим геном в геноме. Если эталонный геном недоступен, теги можно напрямую использовать в качестве диагностических маркеров, если будет обнаружено, что дифференциально экспрессируется в болезненном состоянии.
Экспрессия гена анализа cap (CAGE) метод является вариантом SAGE, который устанавливает последовательность тегов только с 5 'конца транскрипта мРНК. Следовательно, сайт старта транскрипции генов может быть идентифицирован, когда теги выровнены по эталонному геному. Идентификация стартовых сайтов гена используется для анализа промотора и для клонирования полноразмерных кДНК.
Методы SAGE и CAGE позволяют получить информацию о большем количестве генов, чем было возможно при секвенировании отдельных EST, но подготовка образцов и анализ данных обычно более трудозатратны.
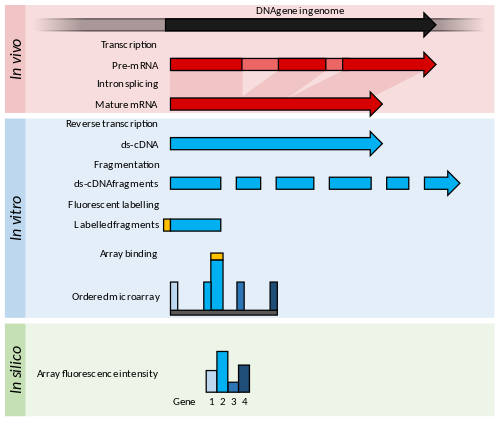 Сводка ДНК-микрочипы. Внутри организмов гены транскрибируются и сплайсируются (у эукариот) с образованием зрелых транскриптов мРНК (красный). МРНК извлекается из организма, и обратная транскриптаза используется для копирования мРНК в стабильную дц-кДНК (синий). В микроматрицах ds-кДНК фрагментирована и флуоресцентно помечена (оранжевый). Меченые фрагменты связываются с упорядоченным массивом комплементарных олигонуклеотидов, и измерение интенсивности флуоресценции по всему массиву указывает на численность заранее определенного набора последовательностей. Эти последовательности обычно специально выбираются для сообщения об интересующих генах в геноме организма.
Сводка ДНК-микрочипы. Внутри организмов гены транскрибируются и сплайсируются (у эукариот) с образованием зрелых транскриптов мРНК (красный). МРНК извлекается из организма, и обратная транскриптаза используется для копирования мРНК в стабильную дц-кДНК (синий). В микроматрицах ds-кДНК фрагментирована и флуоресцентно помечена (оранжевый). Меченые фрагменты связываются с упорядоченным массивом комплементарных олигонуклеотидов, и измерение интенсивности флуоресценции по всему массиву указывает на численность заранее определенного набора последовательностей. Эти последовательности обычно специально выбираются для сообщения об интересующих генах в геноме организма. Микроматрицы состоят из коротких нуклеотидных олигомеров, известных как «зонды ", которые обычно располагаются в виде сетки на предметном стекле. Обилие транскриптов определяют гибридизацией флуоресцентно меченных транскриптов с этими зондами. интенсивность флуоресценции в каждом месте зонда на массиве указывает количество транскриптов для этой последовательности зонда.
Микроматрицы требуют некоторых геномных знаний от интересующего организма, например, в форме аннотированная последовательность генома или библиотека EST, которые можно использовать для создания зондов для массива.
Микроматрицы для транскриптомики обычно попадают в одну из двух широких категорий: точечные массивы с низкой плотностью или массивы с короткими зондами с высокой плотностью. Обилие транскриптов определяется по интенсивности флуоресценции, полученной от меченных флуорофором транскриптов, которые связываются с массивом.
Пятнистые массивы с низкой плотностью обычно содержат пиколитровые капли диапазона очищенного кДНК нанесены на поверхность предметного стекла. Эти зонды длиннее, чем у массивов высокой плотности, и не могут идентифицировать альтернативные события соединения. В пятнистых массивах используются два разных флуорофора для маркировки тестовых и контрольных образцов, а соотношение флуоресценции используется для расчета относительной меры численности. В массивах высокой плотности используется одна флуоресцентная метка, и каждый образец гибридизируется и детектируется индивидуально. Массивы высокой плотности были популяризированы массивом Affymetrix GeneChip, где каждый транскрипт количественно оценивается несколькими короткими 25 -мерными зондами, которые вместе анализируют один ген.
Массивы NimbleGen представляли собой массивы высокой плотности, произведенные методом фотохимии без маски, который позволял гибкое производство массивов в малых или больших количествах. Эти наборы содержали 100000 зондов, содержащих от 45 до 85 элементов, и были гибридизированы с образцом, помеченным одним цветом, для анализа экспрессии. Некоторые дизайны включали до 12 независимых массивов на слайд.
 Краткое изложение RNA-Seq. Внутри организмов гены транскрибируются и сплайсируются (у эукариот) с образованием зрелых транскриптов мРНК (красный). МРНК извлекается из организма, фрагментируется и копируется в стабильную дц-кДНК (синий). Дц-кДНК секвенируют с использованием высокопроизводительных методов секвенирования с коротким считыванием. Эти последовательности затем могут быть выровнены с эталонной последовательностью генома, чтобы реконструировать, какие участки генома транскрибируются. Эти данные можно использовать для аннотации, где находятся экспрессируемые гены, их относительные уровни экспрессии и любые альтернативные варианты сплайсинга.
Краткое изложение RNA-Seq. Внутри организмов гены транскрибируются и сплайсируются (у эукариот) с образованием зрелых транскриптов мРНК (красный). МРНК извлекается из организма, фрагментируется и копируется в стабильную дц-кДНК (синий). Дц-кДНК секвенируют с использованием высокопроизводительных методов секвенирования с коротким считыванием. Эти последовательности затем могут быть выровнены с эталонной последовательностью генома, чтобы реконструировать, какие участки генома транскрибируются. Эти данные можно использовать для аннотации, где находятся экспрессируемые гены, их относительные уровни экспрессии и любые альтернативные варианты сплайсинга. RNA-Seq относится к комбинации высокой производительности Методология секвенирования с помощьювычислительных методов для захвата и количественной оценки транскриптов, присутствующих в экстракте РНК. Генерируемые нуклеотидные ведут обычно длину около 100 п.н., но могут вести себя от 30 п.н. до более 10 000 п.н. в зависимости от используемого метода секвенирования. RNA-Seq использует глубокий выборку транскриптома с множеством коротких фрагментов транскриптома, чтобы обеспечить вычислительную реконструкцию исходного транскрипта РНК путем выравнивания считываний с эталонным геномом или друг с другом (de novo assembly ). РНК как с низким, так и с высоким уровнем качества в эксперименте RNA-Seq (динамический диапазон 5 динамический диапазон ) - преимущество перед транскриптомами микрочипов. Кроме того, количество входящей РНК намного ниже для RNA-Seq (в нанограммах) по сравнению с микипами (количество в микрограммах), что позволяет более тонко исследовать клеточные структуры вплоть до уровня одной клетки в сочетании с линейной амплификацией кДНК. Теоретически не существует верхнего предела количественной оценки в RNA-Seq, а фоновый шум очень низкий для считываний 100 п.н. в неповторяющихся областях.
RNA-Seq может установить определение генов в геном, или определить, какие гены активны в определенный момент времени, а счетчик считываний может быть точного моделирования относительного уровня экспрессии генов. Методология RNA-Seq постоянно совершенствуется, в первую очередь за счет развития технологий секвенирования ДНК для увеличения пропускной способности, точности и длины считывания. С момента первых описаний в 2006 и 2008 годах, RNA-Seq был быстро принят и обогнал микроматрицы в качестве доминирующего метода транскриптомики в 2015 году.
Поиск данных транскриптома на уровне отдельных клеток, привел к прогрессу в области РНК- Методы подготовки библиотеки Seq, приводящие к значительному повышению чувствительности. Транскриптомы отдельных клеток теперь хорошо развиты и даже были расширены до in situ RNA-Seq, где транскриптомы отдельных клеток непосредственно опрашиваются в фиксированных тканях.
RNA-Seq была создана вместе с быстрым развитием ряда технологий высокопроизводительного секвенирования ДНК. Однако перед секвенированием выделенных транскриптов РНК выполняется несколько ключевых этапов обработки. Методы различаются по использованию обогащения транскриптов, фрагментации, амплификации, секвенирования с одним или парным концом, а также по сохранению информации о цепи.
Чувствительность эксперимента RNA-Seq может быть увеличена за счет обогащения классов РНК, которые представляют интерес и истощают известные РНК. Молекулы мРНК можно разделить с помощью олигонуклеотидных зондов, которые связывают свои поли-A-хвосты. В качестве альтернативы, метод гибридизации с зондами, адаптированными к последовательностям рРНК, специфическими для таксона, путем гибридизации с зондами, адаптированными к последовательностям рРНК, специфическим для таксона (например, рРНК млекопитающих, рРНК растений). Однако рибо-истощение может также вносить некоторую систематическую ошибку из-за неспецифического истощения нецелевых транскриптов. Малые РНК, такие как микроРНК, могут быть очищены от зависимости от их размера с помощью гель-электрофореза и экстракции.
мРНК длиннее, чем считывания типичных методов высокопроизводительного секвенирования, транскрипты обычно фрагментируются перед секвенированием. Метод фрагментации - ключевой аспект построения библиотеки секвенирования. Фрагментация может быть достигнута с помощью химического гидролиза, распыления, обработки ультразвуком или обратной транскрипции с цепью -конечные нуклеотиды. В качестве альтернативы, фрагментация и маркировка кДНК могут работать одновременно с использованием ферментов транспозазы.
Во время подготовки к секвенированию копии кДНК транскриптов могут быть амплифицированы с помощью ПЦР для обогащения фрагментов, содержащих ожидаемые 5 ' и 3 'переходные следящие. Амплификация также используется для обеспечения секвенирования очень низких входных количеств РНК, вплоть до 50 пг в экстремальных приложениях. Вспышки контроля Вспышки контроля Известные РНК могут сообщить о качестве контрольная проверка для проверки подготовки библиотеки и секвенирования с точки зрения GC-содержимое, размер фрагмента, а также ущерб из-за положения фрагмента в транскрипте. Уникальные молекулярные механизмы (UMI) предоставляют собой короткие случайные данные, которые используются для индивидуальной пометки фрагментов, используемых во время подготовки библиотеки, чтобы каждый помеченный фрагмент был уникальным. UMI обеспечивает абсолютную шкалу для количественной оценки, возможность корректировать смещение первой амплификации, внесенное во время создания библиотеки, и точно оценивать исходный размер выборки. UMI особенно хорошо подходят для транскриптомики одноклеточной РНК-Seq, где количество входящей РНК ограничено и требуется расширенная амплификация образца.
После получения молекул транскрипта их можно секвенировать в только одно направление (одностороннее) или оба направления (парное). Одноконцевая последовательность обычно получается быстрее, дешевле, чем секвенирование парных концов, и ее для количественной оценки уровней экспрессии генов. Парное секвенирование дает более надежные сопоставления / сборки, что полезно для аннотации генов и обнаружения транскрипта изоформы. Способы, специфичные для цепи RNA-Seq, сохраняют информацию о цепи цепи секвенированного транскрипта. Без информации о цепи считывания могут быть выровнены по локусу гена, но не сообщают, в каком направлении транскрибируется ген. Stranded-RNA-Seq полезен для расшифровки транскрипции генов, перекрывающихся в разных направлениях, и для более надежных прогнозов генов у немодельных организмов.
| Платформа | Коммерческий выпуск | Типичная длина чтения | Максимальная пропускная способность на цикл | Точность однократного чтения | Циклы RNA-Seq, депонированные в NCBI SRA (Октябрь 2016 г.) |
|---|---|---|---|---|---|
| 454 Life Sciences | 2005 | 700 bp | 0,7 Gbp | 99,9% | 3548 |
| Illumina | 2006 | 50–300 bp | 900 Gbp | 99,9 % | 362903 |
| SOLiD | 2008 | 50 бит / с | 320 Гбит / с | 99,9% | 7032 |
| Ion Torrent | 2010 | 400 бит / с | 30 Гбит | 98% | 1953 |
| PacBio | 2011 | 10,000 бит / с | 2 Гбит / с | 87% | 160 |
Условные обозначения: NCBI SRA - Национальный центр биотехнологической информации. Последовательный архив чтения.
В молекуле молекулы кДНК перед секвенированием; следовательно, последующие платформы одинаковы для транскриптомных и геномных данных. Следовательно, развитие технологий секвенирования ДНК было определяющей особенностью RNA-Seq. Прямое секвенирование РНК с использованием секвенирования нанопор представляет собой современный метод RNA-Seq. Секвенирование РНК с помощью нанопор может быть повреждено модифицированные основания, которые в противном случае могут быть замаскированы при секвенировании кДНК, а также исключают этапы амплификации, которые могут внести систематическую ошибку.
Чувствительность и точность Эксперимент RNA-Seq зависит от числа считываний, полученных от каждого образца. Большое количество считываний необходимо для обеспечения достаточного покрытия транскриптома, что позволяет обнаруживать транскрипты с низким уровнем содержания. Дизайн эксперимента дополнительно усложняется технологиями секвенирования с ограниченным диапазоном выходных данных. К этим соображениям применяется разное количество генов и, следовательно, требует индивидуального выхода для эффективного транскриптома. В ранних исследованиях подходящие пороговые значения определялись эмпирически, но по мере развития технологии подходящего покрытия было предсказано расчетным путем по насыщению транскриптома. Несколько нелогично, но наиболее эффективный способ улучшения считываний в генах с низкой экспрессией - это добавить больше биологических реплик, а не добавок больше считываний. Текущие тесты, рекомендованные проект Энциклопедия элементов ДНК (ENCODE), предназначены для 70-кратного покрытия экзома для стандартной системы РНК и до 500-кратного покрытия экзома для обнаружения редких транскриптов и изоформ.
Методы транскриптомики очень параллельны и значительных вычислений для получения значимых данных как для экспериментов с микрочипами, так и для экспериментов с RNA-Seq. Данные микроматрицы записываются в виде изображений с высоким разрешением, требуются признаки обнаружения и спектрального анализа. Размер каждого файла необработанных изображений микрочипа составляет около 750 МБ, а размер обработанных изображений - около 60 МБ. Множественные короткие зонды, соответствующие одному транскрипту, могут раскрыть подробности структуры интрона - экзона, что требует статистических моделей для определения подлинности результирующего сигнала. Исследования RNA-Seq производят миллиарды коротких последовательностей ДНК, которые необходимо выровнять с эталонными геномами, состоящими из миллионов и миллиардов пар оснований. Сборка новых чтений в наборе данных требует построения очень сложных графов последовательностей. Операции RNA-Seq очень часто повторяются и выигрывают от распараллеленных вычислений, но современные алгоритмы означают, что потребительского вычислительного оборудования достаточно для простых экспериментов с транскриптомикой, которые не требуют сборки операций чтения de novo. Человеческий транскриптом может быть точно захвачен с помощью RNA-Seq с 30 миллионами последовательностей по 100 п.н. на образец. В этом случае потребуется 1,8 гигабайта дискового пространства на образце в сжатом fastq. Обработанные данные подсчета для каждого гена будут намного меньше, что эквивалентно мощности обработанных микрочипов. Данные могут следовать в общедоступных репозиториях, таких как Архив чтения (SRA). Наборы данных RNA-Seq можно загрузить через Омнибус экспрессии генов.
 Микроматрица и проточная ячейка секвенирования. Микроматрицы и последовательность РНК по-разному полагаются на анализ изображений. В микроматричном чипе каждый пятно на чипе представляет собой отдельный олигонуклеотидный зонд, а интенсивность флуоресценции напрямую определяет количество конкретной последовательности (Affymetrix). В проточной кювете для высокопроизводительного секвенирования пятна секвенируются по одному нуклеотиду за раз, причем цвет на каждом этапе указывает следующие нуклеотиды в последовательности (Illumina Hiseq). В других вариантах этих методов используется больше или меньше цветовых каналов.
Микроматрица и проточная ячейка секвенирования. Микроматрицы и последовательность РНК по-разному полагаются на анализ изображений. В микроматричном чипе каждый пятно на чипе представляет собой отдельный олигонуклеотидный зонд, а интенсивность флуоресценции напрямую определяет количество конкретной последовательности (Affymetrix). В проточной кювете для высокопроизводительного секвенирования пятна секвенируются по одному нуклеотиду за раз, причем цвет на каждом этапе указывает следующие нуклеотиды в последовательности (Illumina Hiseq). В других вариантах этих методов используется больше или меньше цветовых каналов. Микроматрица обработка изображения должна правильно идентифицировать регулярную сетку функций в изображении и независимо количественно определить интенсивность флуоресценции для каждой функции. Артефакты изображения должны быть идентифицированы и удалены из общего анализа. Интенсивности флуоресценции напрямую на численность каждой последовательной последовательности каждого зонда в массиве известна ужена.
Первые шаги RNA-seq также включают аналогичную обработку изображений; однако преобразование изображений в данные обычно выполняется автоматически программным способом. Метод секвенирования путем введения компании Illumina приводит к созданию кластеров, распределенных по поверхности проточной кюветы. Проточная ячейка визуализируется до четырех раз в течение каждого цикла секвенирования, всего от десятков до сотен циклов. Кластеры проточных кювет аналогичны микрочипов и должны быть правильно идентифицированы на ранних этапах процесса секвенирования. В методе пиросеквенирования Roche 'Интенсивность испускаемого света определяет количество последовательных нуклеотидов в гомополимерном повторе. Существует множество вариантов этих методов, каждый из которых имеет свой профиль ошибок для полученных данных.
Эксперименты RNA-Seq генерируют большой объем считывания необработанных последовательностей, которые имеют для обработки для получения полезной информации. Для анализа данных обычно требуется комбинация инструментов программного обеспечения биоинформатики (см. Также Список инструментов биоинформатики RNA-Seq ), которые различаются в зависимости от плана эксперимента и целей. Процесс можно разбить на четыре этапа: контроль качества, согласование, количественная оценка и дифференциальное выражение. Самые популярные RNA-Seq запускаются из интерфейса командной строки либо в среде Unix, либо в статистической среде R /Bioconductor.
Считывание последовательных последовательностей неидеально, поэтому для последующего анализа необходимо оценивать точность каждой базы в последовательной. Необработанные данные проверяются, чтобы убедиться: оценки качества для базовых вызовов высокие, содержимое GC соответствует ожидаемому распределению, короткие мотивы последовательностей (k-mers ) не представлены чрезмерно, а скорость дублирования чтения приемлемая. низкий. Существует несколько вариантов программного обеспечения для анализа качества последовательности, включая FastQC и FaQC. Аномалии могут быть удалены (обрезаны) или помечены для специальной обработки во время последующих процессов.
Чтобы связать количество считанных последовательностей с экспрессией определенного гена, последовательности транскриптов выровнены с эталонным геномом или выровнены de novo друг к другу, если нет ссылки. Ключевые проблемы для программного обеспечения для выравнивания включают достаточную скорость, позволяющую выровнять миллиарды коротких последовательностей в значимый промежуток времени, гибкость для распознавания и обработки интронного сплайсинга эукариотической мРНК, а также правильное назначение считываний, которые сопоставляются с несколькими локации. Достижения программного обеспечения в значительной степени решают эти проблемы, а увеличение длины последовательного чтения снижает вероятность неоднозначного выравнивания чтения. Список доступных в настоящее время высокопроизводительных выравнивателей последовательностей поддерживается с помощью EBI.
. Выравнивание последовательностей мРНК первичного транскрипта, полученных от эукариот, с эталонным геномом требует специальной обработки интронные последовательности, которые отсутствуют в зрелой мРНК. Выравниватели с коротким считыванием выполняют дополнительный цикл выравниваний, специально предназначенных для идентификации сплайсинговых соединений, на основании канонических последовательностей сайтов сплайсинга и известной информации о сайтах сплайсинга интронов. Идентификация сплайсинговых соединений интрона предотвращает неправильное выравнивание считываний по сплайсинговым стыкам или ошибочное отбрасывание, позволяя выровнять большее количество считываний с эталонным геномом и повышая точность оценок экспрессии генов. Поскольку регуляция гена может происходить на уровне изоформы мРНК, выравнивания с учетом сплайсинга также позволяют обнаруживать изменения численности изоформ, которые в противном случае были бы потеряны при групповом анализе.
Сборка de novo может использоваться для выравнивания считываний друг с другом для создания полноразмерных последовательностей транскриптов без использования эталонного генома. Проблемы, характерные для сборки de novo, включают большие вычислительные требования по сравнению с транскриптомом на основе ссылок, дополнительную проверку вариантов или фрагментов генов и дополнительную аннотацию собранных транскриптов. Первые показатели, используемые для описания сборок транскриптомов, такие как N50, оказались вводящими в заблуждение, и теперь доступны улучшенные методы оценки. Метрики, основанные на аннотациях, позволяют лучше оценить полноту сборки, например, contig обратный счетчик лучших совпадений. После сборки de novo сборку можно использовать в качестве эталона для последующих методов выравнивания последовательностей и количественного анализа экспрессии генов.
| Программное обеспечение | Выпущено | Последнее обновление | Вычислительная эффективность | Сильные и слабые стороны |
|---|---|---|---|---|
| Velvet-Oases | 2008 | 2011 | Низкие, однопоточные, высокие требования к ОЗУ | Исходный ассемблер короткого чтения. Сейчас он в значительной степени заменен. |
| SOAPdenovo-trans | 2011 | 2014 | Умеренные, многопоточные, средние требования к ОЗУ | Ранний пример ассемблера для короткого чтения. Он был обновлен для сборки транскриптома. |
| Trans-ABySS | 2010 | 2016 | Умеренные, многопоточные, средние требования к ОЗУ | Подходит для коротких чтений, может обрабатывать сложные транскриптомы, а для вычислительных кластеров доступна версия MPI-parallel. |
| Trinity | 2011 | 2017 | Умеренные, многопоточные, средние требования к ОЗУ | Подходит для коротких чтений. Он может обрабатывать сложные транскриптомы, но требует большого объема памяти. |
| miraEST | 1999 | 2016 | Умеренные, многопоточные, средние требования к ОЗУ | Может обрабатывать повторяющиеся последовательности, комбинировать различные форматы секвенирования и приемлем широкий диапазон платформ последовательностей. |
| Newbler | 2004 | 2012 | Низкие, однопоточные, высокие требования к ОЗУ | Специализированы для устранения ошибок гомополимерного секвенирования, типичных для Секвенсоры Roche 454. |
| CLC genomics workbench | 2008 | 2014 | Высокие, многопоточные, низкие требования к ОЗУ | Имеет графический пользовательский интерфейс, может сочетать различные технологии секвенирования, не имеет специфичных для транскриптомов функций, и перед использованием необходимо приобрести лицензию. |
| SPAdes | 2012 | 2017 | Высокие, многопоточные, низкие требования к ОЗУ | Используется для экспериментов по транскриптомике на отдельных клетках. |
| RSEM | 2011 | 2017 | Высокие, многопоточные, низкие требования к ОЗУ | Может оценить частоту альтернативно соединенных транскриптов. Дружественный интерфейс. |
| StringTie | 2015 | 2019 | Высокие, многопоточные, низкие требования к ОЗУ | Можно использовать комбинацию ориентированного на ссылку и новые методы сборки для идентификации транскриптов. |
Условные обозначения: RAM - оперативная память; MPI - интерфейс передачи сообщений; EST - тег экспрессируемой следовать.
Количественная оценка выравнивания последовательностей может быть выполнена на уровне гена, экзона или транскрипта. Типичные выходные данные включают в себя таблицу счетчиков чтения для каждой функции, предоставленной программному обеспечению; например, для генов в файле общего формата признаков. Подсчет считывания генов и экзонов может быть довольно легко рассчитан, например, с помощью HTSeq. Количественный анализ на уровне транскрипта более сложен и требует вероятностных методов для оценки количества изоформ транскрипта на основе короткой информации чтения; например, с помощью программного обеспечения для запонок. Считывания, которые одинаково хорошо совпадают с несколькими местоположениями, должны быть идентифицированы и либо удалены, либо выровнены по одному из возможных местоположений, либо выровнены по наиболее вероятному местоположению.
Некоторые методы количественной оценки могут полностью обойти необходимость в точном выравнивании считываемого изображения с эталонной последовательностью. Программный метод kallisto объединяет псевдо-выравнивание и количественную оценку в один этап, который выполняется на 2 порядка быстрее, чем современные методы, такие как те, которые используются в программном обеспечении tophat / cufflinks, с меньшей вычислительной нагрузкой.
Когда доступны количественные подсчеты каждого транскрипта, дифференциальная экспрессия гена измеряется путем нормализации, моделирования и статистического анализа данных. Большинство инструментов будут считывать таблицу генов и считывать счетчики в качестве входных данных, но некоторые программы, такие как cuffdiff, принимают в качестве входных данных выравнивания чтения в формате карты двоичного выравнивания. Конечными результатами этих анализов являются списки генов с соответствующими попарными тестами для дифференциальной экспрессии между видами лечения и оценки вероятности этих различий.
| Программное обеспечение | Среда | Специализация |
|---|---|---|
| Cuffdiff2 | Unix-based | Анализ транскриптов, отслеживающий альтернативный сплайсинг мРНК |
| EdgeR | R / Bioconductor | Любые геномные данные на основе подсчета |
| DEseq2 | R / Bioconductor | Гибкие типы данных, низкая репликация |
| Limma/Voom | R / Bioconductor | Данные микроматрицы или РНК-Seq, гибкий дизайн эксперимента |
| Ballgown | R / Bioconductor | Эффективное и чувствительное обнаружение транскриптов, гибкость. |
Обозначения: мРНК - информационная РНК.
Транскриптомный анализ может быть подтвержден с использованием независимого метода, например, количественной ПЦР (qPCR), которая распознается и статистически поддающийся оценке. Экспрессию генов измеряют относительно определенных стандартов как для интересующего гена, так и для генов контрольных. Измерение с помощью qPCR аналогично измерению, полученному с помощью RNA-Seq, где значение может быть рассчитано для концентрации целевой области в данном образце. Однако qPCR ограничивается ампликонами размером менее 300 п.н., обычно ближе к 3 ’концу кодирующей области, избегая 3’UTR. Если требуется проверка изоформ транскриптов, проверка выравниваний чтения RNA-Seq должна указать, где qPCR праймеры могут быть размещены для максимальной дискриминации. Измерение нескольких контрольных генов вместе с интересующими генами дает стабильный эталон в биологическом контексте. Проверка данных РНК-Seq методом qPCR в целом показала, что различные методы RNA-Seq сильно коррелированы.
Функциональная проверка ключевых генов является важным фактором при посттранскриптомном планировании. Наблюдаемые паттерны экспрессии генов могут быть функционально связаны с фенотипом с помощью независимого нокдауна / спасательного исследования в интересующем организме.
Транскриптомные стратегии нашли широкое применение в различных областях биомедицинских исследований, включая диагностику и профилирование. Подходы RNA-Seq позволили провести крупномасштабную идентификацию стартовых сайтов транскрипции, выявить использование альтернативного промотора и новые изменения сплайсинга. Эти регуляторные элементы важны при заболевании человека, и поэтому определение таких вариантов имеет решающее значение для интерпретации исследований ассоциации заболеваний. RNA-Seq также может идентифицировать связанные с заболеванием однонуклеотидные полиморфизмы (SNP), аллель-специфическую экспрессию и слияния генов, что способствует пониманию причинных вариантов заболевания.
Ретротранспозоны представляют собой мобильные элементы, которые размножаются в геномах эукариот посредством процесса, включающего обратную транскрипцию. RNA-Seq может предоставить информацию о транскрипции эндогенных ретротранспозонов, которые могут влиять на транскрипцию соседних генов посредством различных эпигенетических механизмов, которые приводят к заболеванию. Точно так же потенциал использования RNA-Seq для понимания заболевания, связанного с иммунитетом, быстро расширяется благодаря способности анализировать популяции иммунных клеток и упорядочивать Т-клетки и В-клетки. рецепторы от пациентов.
RNA-Seq человеческих патогенов стали общепринятым методом количественной оценки изменений экспрессии генов, выявляя новые факторы вирулентности, прогнозирование устойчивости к антибиотикам и выявление иммунных взаимодействий между хозяином и патогеном. Основная цель этой технологии - разработать оптимизированные меры инфекционного контроля и целевое индивидуализированное лечение.
Транскриптомный анализ преимущественно сосредоточен либо на хозяине, либо на патогене. Dual RNA-Seq применялся для одновременного профилирования экспрессии РНК как у патогена, так и у хозяина на протяжении всего процесса инфицирования. Этот метод позволяет изучать динамический ответ и межвидовые сети регуляции генов у обоих партнеров по взаимодействию от начального контакта до инвазии и окончательной персистенции патогена или его удаления иммунной системой хозяина.
Транскриптомика позволяет идентифицировать гены и пути, которые реагируют на биотические и абиотические стрессы окружающей среды и противодействуют им. Нецелевые Природа транскриптомики позволяет идентифицировать новые транскрипционные сети в сложных системах. Например, сравнительный анализ ряда линий нута на разных стадиях развития выявил различные профили транскрипции, связанные со стрессами засухи и засоления, включая определение роли изоформы транскрипта из AP2 - EREBP. Исследование экспрессии генов во время формирования биопленки с помощью грибкового патогена Candida albicans выявило ко-регулируемый набор генов, критических для образования и поддержания биопленки.
Транскриптомное профилирование также предоставляет важную информацию о механизмах лекарственной устойчивости. Анализ более 1000 изолятов Plasmodium falciparum, вирулентного паразита, ответственного за малярию у людей, выявил повышенную регуляцию развернутого белкового ответа и более медленное прогрессирование на ранних стадиях бесполого внутриэритроцитарного цикл развития были связаны с устойчивостью к артемизинину в изолятах из Юго-Восточной Азии.
Все транскриптомные методы были особенно полезны для идентификации функции генов и выявление тех, кто отвечает за определенные фенотипы. Транскриптомика арабидопсиса экотипов, которые гипераккумулируют металлы, коррелируют гены, участвующие в поглощении металлов, толерантности и гомеостазе с фенотипом. Интеграция наборов данных RNA-Seq в различных тканях использовалась для улучшения аннотации функций генов у коммерчески важных организмов (например, огурец ) или исчезающих видов (например, коала ).
Сборка RNA-Seq читает не зависит от эталонного генома и поэтому идеально подходит для исследований экспрессии генов немодельных организмов с несуществующими или плохо развитыми геномными ресурсами. Например, база данных SNP, используемая в пихте Дугласа программы разведения были созданы с помощью анализа транскриптомов de novo в отсутствие секвенированного генома. Точно так же гены, которые участвуют в развитии сердечной, мышечной и нервной ткани у лобстеров, были идентифицированы путем сравнения транскриптомов различных типов тканей без использования последовательности генома. RNA-Seq также может использоваться для идентификации ранее неизвестных областей кодирования белка в существующих секвенированных геномах.
Профилактика старения Все это невозможно без измерения скорости старения. Самый современный и сложный способ измерения скорости старения - это использование различных биомаркеров старения человека, основанный на использовании глубоких нейронных сетей, которые могут быть обучены на любом типе биологических данных омики для прогнозирования возраста субъекта. Было показано, что старение является сильной движущей силой изменений транскриптома. Часы старения, основанные на транскриптомах, страдали от значительного разброса данных и относительно низкой точности. Однако подход, который использует временное масштабирование и бинаризацию транскриптомов для определения набора генов, который предсказывает биологический возраст с точностью, позволил достичь оценки, близкой к теоретическому пределу.
Транскриптомика чаще всего применяется к содержанию мРНК клетки. Однако те же методы в равной степени применимы к некодирующим РНК (нкРНК), которые не транслируются в белок, но вместо этого выполняют прямые функции (например, роли в трансляции белка, репликации ДНК, сплайсинг РНК и регуляция транскрипции ). Многие из этих нкРНК влияют на болезненные состояния, включая рак, сердечно-сосудистые и неврологические заболевания.
Исследования транскриптомики генерируют большие объемы данных, которые имеют потенциальное применение, выходящее далеко за рамки первоначальных целей эксперимента.. Таким образом, необработанные или обработанные данные могут быть депонированы в общедоступных базах данных, чтобы обеспечить их полезность для более широкого научного сообщества. Например, по состоянию на 2018 год Омнибус экспрессии генов содержал миллионы экспериментов.
| Имя | Хост | Данные | Описание |
|---|---|---|---|
| Ген Expression Omnibus | NCBI | Microarray RNA-Seq | Первая база данных транскриптомики для приема данных из любого источника. Введены стандарты сообщества MIAME и MINSEQE, которые определяют необходимые метаданные эксперимента для обеспечения эффективной интерпретации и повторяемости. |
| ArrayExpress | ENA | Microarray | Импортирует наборы данных из Омнибуса экспрессии генов и принимает прямую отправку. Обработанные данные и метаданные эксперимента хранятся в ArrayExpress, а считывания необработанной последовательности - в ENA. Соответствует стандартам MIAME и MINSEQE. |
| Атлас экспрессии | EBI | Microarray RNA-Seq | База данных тканеспецифической экспрессии генов для животных и растений. Отображает вторичный анализ и визуализацию, например, функциональное обогащение терминов Gene Ontology, доменов InterPro или путей. Ссылки на данные о содержании белка, если таковые имеются. |
| Genevestigator | Частный курируемый | Microarray RNA-Seq | Содержит ручные курирования общедоступных наборов данных транскриптомов с упором на медицинские данные и данные биологии растений. Индивидуальные эксперименты нормализованы по всей базе данных, что позволяет сравнивать экспрессию генов в различных экспериментах. Полная функциональность требует покупки лицензии с бесплатным доступом к ограниченной функциональности. |
| RefEx | DDBJ | Все | Транскриптомы человека, мыши и крысы из 40 различных органов. Экспрессия генов визуализировалась как тепловые карты, спроецированные на трехмерные изображения анатомических структур. |
| NONCODE | noncode.org | RNA-Seq | Некодирующие РНК (нкРНК), за исключением тРНК и рРНК. |
Легенда: NCBI - Национальный центр биотехнологической информации; EBI - Европейский институт биоинформатики; DDBJ - Банк данных ДНК Японии; ENA - Европейский архив нуклеотидов; MIAME - Минимум информации об эксперименте с микрочипом; MINSEQE - Минимум информации об эксперименте по SEQuencing с высокой пропускной способностью.
![]() Эта статья была адаптирована из следующего источника под лицензия CC BY 4.0 () (отчеты рецензента ): Рохан Лоу; Нил Ширли; Марк Блекли; Стивен Долан; Томас Шафи (2017), «Технологии транскриптомики», PLOS Computational Biology, 13 (5): e1005457, doi : 10.1371 / JOURNAL.PCBI.1005457, PMC 5436640, PMID 28545146, Wikidata Q33703532
Эта статья была адаптирована из следующего источника под лицензия CC BY 4.0 () (отчеты рецензента ): Рохан Лоу; Нил Ширли; Марк Блекли; Стивен Долан; Томас Шафи (2017), «Технологии транскриптомики», PLOS Computational Biology, 13 (5): e1005457, doi : 10.1371 / JOURNAL.PCBI.1005457, PMC 5436640, PMID 28545146, Wikidata Q33703532