
Войти
В биоинформатике, a выравнивание последовательностей - это способ упорядочивания последовательностей , РНК или ДНК могут использовать сходства, которые могут быть следствием функциональных, структурных или эволюционные отношения между связями. Выровненные остатки следующих нуклеотидных или аминокислот обычно представляют в виде строк в матрице. Между остатками вставляются пробелы, одинаковые или похожие символы были выровнены в последовательных столбцах. Выравнивание последовательностей также используется для небиологических последовательностей, например для расчета расстояния между строками на языковом языке или в финансовых данных.
 Выравнивание последовательностей, продуцируемых ClustalO, белков гистонов млекопитающих.. Последовательности представьте собой аминокислоты для остатков 120-180 белков. Остатки, которые сохраняются во всех последовательностях, выделены серым цветом. Ниже белковых последовательностей находится ключ, обозначающий консервативную последовательность (*), консервативные мутации (:), полуконсервативные мутации (.) И неконсервативные мутации ().
Выравнивание последовательностей, продуцируемых ClustalO, белков гистонов млекопитающих.. Последовательности представьте собой аминокислоты для остатков 120-180 белков. Остатки, которые сохраняются во всех последовательностях, выделены серым цветом. Ниже белковых последовательностей находится ключ, обозначающий консервативную последовательность (*), консервативные мутации (:), полуконсервативные мутации (.) И неконсервативные мутации (). Если две следуют в одном выравнивании разделяют ком предка, несоответствия могут быть интерпретированы как точе мутации, а пробелы как indels (то есть инсерционные или делеционные мутации), введенные в одну или обе линии в то время, когда они расходились друг от друга. При выравнивании последовательностей белков степень сходства между аминокислотами, занимают конкретное положение в последовательности, можно интерпретировать как грубую меру того, насколько консервативно конкретная область или последовательность. мотив входит в число родословных. Отсутствие или наличие только очень консервативных замен (то есть замена замен, чьи боковые цепи имеют значение аналогичные биохимические свойства) в конкретной области предполагают, что эта область имеет структурные или функциональные свойства. ДНК и РНК нуклеотидные основания больше похожи друг на друга, чем аминокислоты, сохранение оснований может указывать на функциональную функциональную роль.
Очень короткие или очень похожие следовать можно выровнять вручную. Тем не менее, требуют выравнивания длинных, сильно вариабельных или самых интересных последовательностей, которые могут быть выровнены исключительно усилиями человека. Вместо этого человеческие знания применяются при построении алгоритмов для получения высококачественных выравниваний последовательностей, а иногда и при корректировке окончательных результатов для отражения алгоритмов, которые сложно представить алгоритмически (особенно в случае нуклеотидных последовательностей). Вычислительные подходы к выравниванию последовательностей обычно делятся на две категории: глобальное выравнивание и локальные выравнивания. Вычисление глобального выравнивания - это форма глобальной оптимизации, которая заставляет выравнивание охватывать всю совокупность всех запрашиваемых последовательностей. Напротив, локальные выравнивания идентифицируют области сходства в длинных последовательностях, которые в целом сильно расходятся. Часто предпочтительнее локальное выравнивание, но его бывает труднее вычислить из дополнительной проблемы, нет с идентификацией области сходства. К задаче выравнивания последовательностей были применены вычислительные алгоритмы. К ним относятся медленные, но формально правильные методы, такие как динамическое программирование. К ним также относятся эффективные эвристические алгоритмы или вероятностные методы, разработанные для крупномасштабного поиска в базе данных, которые не гарантируют нахождение наилучших совпадений.
Ref. : GTCGTAGAATA. Чтение : CACGTAG - TA. CIGAR: 2S5M2D2M.
где:. 2S = 2 несоответствия. 5M = 5 совпадений. 2D = 2 удаления. 2M = 2 совпадения.
. Выравнивания обычно как графически, так и в текстовом формате. Почти во всех представлениях выравнивания последовательных последовательностей записываются в ряды последовательности последовательностей, чтобы выровненные последовательности выполнялись в последовательных столбцах. В текстовых форматах выровненные столбцы, содержащие одинаковые или похожие символы, обозначаются систему значений. Как и на изображении выше, звездочка или вертикальная черта используются для обозначения идентичности между двумя столбцами; другие менее распространенные символы включают двоеточие для консервативных замен и точку для полуконсервативных замен. Многие визуализации показывают также используются цвет для отображения информации о свойствах отдельных элементов программы; в последовательности ДНК и РНК это равносильно присвоению каждому нуклеотиду своего собственного цвета. При выравнивании белков, например, на изображении выше, цвет часто используется для обозначения свойств аминокислот, чтобы помочь в оценке консервации замены аминокислот. Для нескольких последовательностей последняя строка в каждом столбце часто является консенсусной последовательностью , определенным выравниванием; консенсусная последовательность также представлена в графическом формате с логотипом снабжения, в котором размер каждой буквы нуклеотида или аминокислоты соответствует степени ее сохранения.
Выравнивания последовательности можно в широком спектре текстовых форматов, многие из которых изначально были разработаны в программе связи или реализации выравнивания. Большинство веб-инструментов допускают ограниченное количество форматов ввода и вывода, таких как формат FASTA и формат GenBank, и вывод не легко редактируется. Доступно несколько программ преобразования, которые используют графический интерфейс и / или интерфейс командной строки, например READSEQ и EMBOSS. Существует также несколько программных пакетов, которые обеспечивают эту функцию преобразования, например, BioPython, BioRuby и BioPerl. В файлах SAM / BAM используется строковый формат CIGAR (Compact Idiosyncratic Gapped Alignment Report) для представления выравнивания относительно путем последовательного кодирования событий (например, совпадение / несоответствие, вставки, удаление).
Глобальные выравнивания, которые пытаются выровнять каждый остаток в каждой последовательности, наиболее полезны, когда в наборе запроса схожи и примерно равного размера. (Это не означает, что глобальное выравнивание может начинаться / или заканчиваться пробелами.) Общим методом глобального выравнивания алгоритм Нидлмана - Вунша, который основан на динамическом программировании. Локальное выравнивание более полезно для разнородных последовательностей, которые, как основные положения, содержат положения, исходящие из их более крупных последовательностей. Алгоритм Смита - Уотермана - это общий метод локального выравнивания, основанный на той же схеме динамического программирования, но с дополнительными вариантами начала и конца в любом месте.
Гибридные методы, известные как полуглобальные или "глокальные" (сокращение от glo bal-lo cal ) методы, поиск наилучшего возможного частичного выравнивания двух последовательностей (другими словами, комбинация одного или обоих запусков заявлено, что один или оба конца выровнены). Это может быть особенно полезно, когда нисходящая часть одной заставляется с восходящей другой другой монтаж. В этом случае ни глобальное выравнивание не является полностью подходящим: глобальное выравнивание будет пытаться выравнивать выход за пределы области перекрытия, в то время как локальное выравнивание может не полностью покрывать область перекрытия. Другой случай, когда полезно полуглобальное выравнивание, - это когда одна последовательность короткая (например, последовательность гена), а другая очень длинная (например, последовательность хромосомы). В этом случае короткая последовательность должна быть глобально (полностью) выровнена, но для последовательной последовательности желательно только локальное (частичное) выравнивание.
Быстрое распространение генетических данных ставит под сомнение текущих алгоритмов выравнивания последовательностей ДНК. Существенные потребности в эффективном и точном методе обнаружения ДНК требуют инновационных подходов для параллельной обработки в реальном времени. Оптические вычисления были предложены в качестве многообещающей альтернативы существующим реализацией, но их применимость еще предстоит проверить [1].
Способы попарного выравнивания последовательности используются для наиболее подходящих кусочных (локальных или глобальных) выравниваний двух последовательностей запроса. Попарное выравнивание можно использовать одновременно между двумя системами, но они эффективны для вычислений и часто используются для методов, не требующих высокой точности (таких как поиск в базе последовательностей с высоким сходством с запросом). Три основных метода обработки парных выравниваний - методы динамического программирования и методы слов; однако методы множественного выравнивания последовательностей также могут выравнивать пары последовательностей. Хотя каждый метод имеет свои сильные и слабые стороны, все несколько повторяющихся последовательностей с низким уровнем информационным содержанием - особенно там, где количество повторений отличается в двух последовательностях, которые выровнять. Одним из способов количественной оценки полезности данного попарного выравнивания является «максимальное уникальное совпадение» (MUM) или самая длинная подпоследовательность, которая встречается в любых запросных последовательностях. Более длинные следовать MUM обычно отражают более близкое родство.
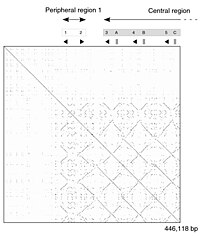 Самостоятельное сравнение части генома линии мыши. Точечный график показывает мозаику из линий, демонстрирующих дублированные сегменты ДНК. Самостоятельное сравнение части генома линии мыши. Точечный график показывает мозаику из линий, демонстрирующих дублированные сегменты ДНК. |
 Точечный график ДНК транскрипции человека цинковый палец коэффициент (идентификатор GenBank NM_002383), показывающий региональное самоподобие. Основная диагональ представляет собой выравнивание с самой собой; линии от главной диагонали соответствуют себе или повторяющиеся узоры в последовательность. Это типичный пример графика повторения. Точечный график ДНК транскрипции человека цинковый палец коэффициент (идентификатор GenBank NM_002383), показывающий региональное самоподобие. Основная диагональ представляет собой выравнивание с самой собой; линии от главной диагонали соответствуют себе или повторяющиеся узоры в последовательность. Это типичный пример графика повторения. |
Точечно-матричный подход, который неявно создает семейство выравниваний для отдельных последовательностей, качественно и концептуально прост, хотя требует времени для анализа в крупном масштабе. В отсутствие шума можно легко идентифицировать внедренные - такие как вставки, удаление, повторы или инвертированные повторы - на точечно-матричный графике. Чтобы построить точечно-матричный график , две последовательности приведены вдоль верхней строки и крайнего левого столбца двумерной матрицы, а точка помещается в любую точку, где символы в соответствии соответствующих столбцов - это типичный график повторения . Некоторые изменяют реализацию или интенсивность реализации в зависимости от степени сходства двух символов, чтобы обеспечить консервативные замены. Точечные диаграммы связанных последовательностей в виде одной линии вдоль главной диагонали матрицы.
. Проблемы с точечными графиками в качестве метода вывода информации включают: шум, нечеткость, неинтуитивность, сложность извлечения сводки статистики и совпадение позиций по двум последовательностям. Также много бесполезного места, где данные по своей природе дублируются по диагонали, и большая часть фактической области работает либо пустым пространством, либо шумом, и, наконец, точечные графики ограничены двумя последовательностями. Ни одно из этих ограничений не распространяется на диаграммы выравнивания Miropeats, но у них есть свои особые недостатки.
Точечные диаграммы также Заявление о соблюдении одной из установленных. Последовательность может быть нанесена на графике против самой, и области, имеющей большое сходство в виде линий от главной диагонали. Этот эффект может возникнуть, когда белок состоит из нескольких похожих структурных доменов.
Техника динамического программирования может быть обеспечение глобального выравнивания через Алгоритм Нидлмана-Вунша и локальные выравнивания с помощью алгоритма Смита-Уотермана. В типичном использовании при выравнивании белков используется матрица замен для присвоения балловых совпадений или несоответствиям аминокислот и штраф за пробел за соответствие аминокислотам в одной последовательности пробелу в Другие. Для выравнивания ДНК и РНК использовать матрицу оценок. (В стандартном динамическом программировании оценки положения аминокислот не зависит от идентичности ее соседей, и поэтому эффекты укладки оснований не принимаются во внимание. Такие эффекты можно учесть с помощью изменения алгоритма.) Распространенным расширенным является стандартная стоимость линейного разрыва. использование двух различных штрафов за разрыв для открытия разрыва и для увеличения разрыва. Обычно первый намного больше второго, например -10 для открытия гэпа и -2 для расширения гэпа. Таким образом, количество пробелов в выравнивании обычно уменьшается, а остатки и пробелы сохраняются вместе, что обычно имеет больший биологический смысл. Алгоритм Гото реализует затраты на аффинный разрыв с помощью трех матриц.
Динамическое программирование может быть полезно при выравнивании нуклеотидных последовательностей с белковыми последовательностями, задача, усложняемая необходимостью соотношения мутации сдвиговые рамки считывания (обычно вставки или делеции). Метод поиска по кадрам производит серию глобальных или локальных попарных выравниваний между запрашиваемой нуклеотидной последовательностью и поисковым набором последовательностей белков или наоборот. Его способность оценивать сдвиг кадров, смещенный произвольным число нуклеотидов, делает этот метод для последовательности последовательностей, делает большое количество отступов, может быть очень трудно согласовать более эффективными эвристическими методами. На практике для этого метода требуются большие вычислительные мощности или система, архитектура специализирована для динамического программирования. Наборы BLAST и EMBOSS предоставляют базовые инструменты для создания транслированных выравниваний (хотя некоторые из этих подходов используют преимущества побочных эффектов, возможности поиска последовательностей этих инструментов). Более общие методы доступны из программного обеспечения с открытым исходным кодом, такого как GeneWise.
. Метод динамического программирования гарантированно найдет оптимальное выравнивание с учетом конкретной оценочной функции; однако оценочной функции часто является эмпирическим, а не теоретическим вопросом. Хотя динамическое программирование расширяется до более чем двух последовательностей, оно недопустимо медленное для большого количества последовательностей или очень длинных последовательностей.
Методы Word, также известные как методы k-tuple, являются эвристическими методами, которые не гарантируют нахождение оптимального решения для выравнивания, но значительно более эффективны чем динамическое программирование. Эти методы особенно полезны при крупномасштабном поиске в базе данных, когда понятно, что большая часть последовательностей-кандидатов не будет иметь существенного совпадения с последовательностью запроса. Методы Word наиболее известны своей реализацией в инструментах поиска в базе данных FASTA и в семействе BLAST. Методы Word идентифицируют серию коротких неперекрывающихся подпоследовательностей («слов») в последовательности запроса, которые затем сопоставляются с последовательностями базы данных кандидатов. Относительные позиции слова в двух сравниваемых последовательностях вычитаются для получения смещения; это укажет на область выравнивания, если несколько отдельных слов производят одинаковое смещение. Только если эта область обнаружена, эти методы применяют более чувствительные критерии выравнивания; таким образом, устраняются многие ненужные сравнения с последовательностями, не имеющими заметного сходства.
В методе FASTA пользователь определяет значение k, которое будет использоваться в качестве длины слова для поиска в базе данных. Этот метод медленнее, но более чувствителен при более низких значениях k, которые также предпочтительны для поиска, включающего очень короткую последовательность запросов. Семейство методов поиска BLAST предоставляет ряд алгоритмов, оптимизированных для определенных типов запросов, таких как поиск отдаленно связанных совпадений последовательностей. BLAST был разработан, чтобы предоставить более быструю альтернативу FASTA без ущерба для точности; как и FASTA, BLAST использует поиск слова длины k, но оценивает только совпадения наиболее значимых слов, а не каждое совпадение слов, как FASTA. Большинство реализаций BLAST используют фиксированную длину слова по умолчанию, которая оптимизирована для запроса и типа базы данных и изменяется только при особых обстоятельствах, например, при поиске с повторяющимися или очень короткими последовательностями запросов. Реализации можно найти через ряд веб-порталов, таких как EMBL FASTA и NCBI BLAST.
 Выравнивание 27 птичьего гриппа белковые последовательности гемагглютинина, окрашенные в соответствии с консервацией остатка (вверху) и свойствами остатка (внизу)
Выравнивание 27 птичьего гриппа белковые последовательности гемагглютинина, окрашенные в соответствии с консервацией остатка (вверху) и свойствами остатка (внизу) Множественное выравнивание последовательностей - это расширение попарного выравнивания для включения более двух последовательностей за раз. Множественные методы выравнивания пытаются выровнять все последовательности в заданном наборе запросов. Множественные выравнивания часто используются для идентификации областей консервативных последовательностей в группе последовательностей, предположительно связанных эволюционно. Такие консервативные мотивы последовательности можно использовать в сочетании со структурной и механистической информацией для определения местоположения каталитических активных сайтов ферментов. Выравнивания также используются для помощи в установлении эволюционных отношений путем построения филогенетических деревьев. Множественные выравнивания последовательностей сложно произвести с вычислительной точки зрения, и большинство формулировок проблемы приводят к NP-полным задачам комбинаторной оптимизации. Тем не менее, использование этих выравниваний в биоинформатике привело к развитию множества методов,подходящих для выравнивания трех или более последовательностей.
Техника динамического программирования теоретически применима к количеству последовательностей; однако, поскольку он требует больших затрат времени и памяти, он редко используется для более чем трех или четырех последовательностей в его самой простой форме. Этот метод требует построения n-мерного эквивалента матрицы последовательностей, сформированной из двух последовательностей, где n - количество последовательностей в запросе. Стандартное динамическое программирование сначала используется для всех пар запрашиваемых последовательностей, а затем «пространство для выравнивания» заполняется попытка совпадений или пробелов в промежуточных положениях, в конечном итоге создается выравнивание между каждым выравниванием двух последовательностей. Хотя этот метод является дорогостоящим с точки зрения вычислений, его гарантия глобального оптимального решения полезна в случаях, когда необходимо точно выровнять только несколько последовательностей. В программном пакете MSA был реализован один метод сжатия функций динамического программирования, основанный на «сумме пар» задача.
Прогрессивные, иерархические или древовидные методы генерируют множественное выравнивание последовательностей, сначала выравнивая наиболее похожие последовательности, а также добавляя следующие соответствующие последовательности или группы к выравниванию до тех пор, пока весь набор запросов не будет включен в решение. Первоначальное дерево, описывающее родство последовательностей, основанные на методах попарного выравнивания, подобных FASTA. Результаты прогрессивного выравнивания зависят от выбора «наиболее родственных» последовательностей и, таким образом, чувствительны к неточности в начальных парных выравниваниях. Большинство методов прогрессивного множественного выравнивания последовательностей дополнительно выполняют в наборе запроса в соответствии с их родством, что снижает вероятность неправильного исходных последовательностей и, таким образом, повышает точность выравнивания.
Многие варианты прогрессивной реализации Clustal используются для множественного выравнивания последовательностей, построения филогенетического дерева и в качестве входных данных для предсказания структуры белка. Более медленный, но более точный вариант прогрессивного метода как T-Coffee.
Итерационные методы улучшают сильную зависимость от точности начальных парных сопоставлений, которая является слабым местом прогрессивных методов. Итерационные методы оптимизируют целевую функцию на основе выбранного метода оценки выравнивания, назначая начальное глобальное выравнивание и повторно выравнивая подмножества последовательностей. Затем повторно выровненные подмножества сами выравниваются, чтобы произвести множественное выравнивание последовательностей в следующей итерации. Рассмотрены различные способы выбора подгрупп последовательностей и целевые функции.
Поиск мотивов, также известный как анализ профиля, конструирует глобальные множественные выравнивания последовательностей, которые пытаются выровнять короткие консервативные мотивы последовательностей последовательностей в наборе запроса. Обычно это делается путем сначала построения глобального множественного выравнивания последовательностей, после чего высоко консервативные области используют для построения набора матриц профилей. Матрица профиля для каждой консервативной области устроена как матрица оценок, но ее частотные подсчеты для каждой аминокислоты или нуклеотида в каждом положении выводятся из распределения признаков консервативной области, а не из более общего эмпирического распределения. Затем матрицы профилей используются для поиска в других схемах поиска мотивации. В случаях, когда исходный набор данных содержал небольшие последовательности или только сильно родственные данные, добавляются псевдосчетчики для нормы распределения символов, представленных в мотиве.
 Профиль HMM, моделирующий множественное выравнивание последовательностей
Профиль HMM, моделирующий множественное выравнивание последовательностей К множественным последовательностям также были применены общие оптимизационные алгоритмы, обычно используемые в информатике. проблема выравнивания. Скрытые марковские модели использовались для получения оценок вероятности для системы возможных выравниваний последовательностей для данного набора запросов; Хотя ранние методы на основе HMM давали невысокую производительность, более поздние приложения их особенно эффективными при обнаружении удаленных последовательностей, поскольку они подвержены шуму, создаваемому консервативными или полуконсервативными заменами. также использовались при оптимизации множественного выравнивания последовательностей, чем судили с помощью функций оценки, такой как метод суммы пар. Более полные сведения и программные пакеты можно найти в основной статье выравнивание множественных последовательностей.
преобразование Барроуза - Уиллера было успешно применено для быстрого выравнивания короткого чтения в популярных инструментах, таких как Bowtie и BWA. См. FM-индекс.
Структурное выравнивание, которое обычно специфично для последовательностей белка, а иногда и РНК, использует информацию о вторичной и третичной структуре молекулы белка или РНК, чтобы помочь в выравнивании последовательностей. Эти методы можно использовать для двух или более последовательностей и обычно дают локальное выравнивание; однако, они зависят от наличия структурной информации, их можно использовать только для последовательностей, соответствующие структуры известны (обычно с помощью рентгеновской кристаллографии или ЯМР-спектроскопии ). Сравнение последовательностей не может надежно их сходство, которые очень отдаленно связаны друг с другом.
Структурные выравнивания используются в качестве «золотого стандарта» при выравнивании для основанного на гомологии предсказания структуры белка. о следовать. Однако не используется механизм прогнозирования структуры. Было показано, что при структурном выравнивании между последовательностью-мишенью и последовательностью-матрицей можно получить высокоточные модели последовательности целевого белка; Основное камнем преткновения в прогнозировании структуры на основе гомологии.
Метод DALI или матрица расстояний выравнивание, представляет собой метод на основе фрагментов построения структурных выравниваний на основе шаблоновного сходства между последовательностями гексапептидов в запрашиваемых последовательностях. Он может генерировать попарные или множественные выравнивания и идентифицировать структурных соседей запрашиваемую последовательность в Protein Data Bank (PDB). Его использовали для создания базы данных структурного выравнивания FSSP (складчатая классификация, основанная на структурном выравнивании белков или семейств структурно подобных белков). Доступ к веб-серверу DALI можно получить по адресу DALI, а FSSP находится по адресу База данных Dali.
SSAP (программа последовательного выравнивания структуры) - это метод, основанный на динамическом программировании. структурного выравнивания, которое в качестве сравнения используются атомно-атомные структуры в структуре выравнивания. Он был расширен с момента своего первоначального описания для включения множественных, а также попарных выравниваний и использовался при построении иерархической классификации белковых складок CATH (класс, архитектура, топология, гомология) в базе данных. Доступ к базе данных CATH можно по адресу Классификация структурных структур CATH.
Метод комбинаторного расширения структурного выравнивания генерирует попарное структурное выравнивание с использованием структуры геометрии для выравнивания коротких фрагментов двух белков анализируется, собирает эти фрагменты в более крупный ряд. Основываясь на таких показателях, как твердое тело среднеквадратичное расстояние, расстояния между остатками, локальная вторичная структура и особенности окружающей среды, такие как соседний остаток гидрофобность, локальные выравнивания называются «выровненными парами фрагментами». и используются для матрицы подобия, представляющие все возможные структурные совпадения в определенных заранее критериев отсечения. Затем путь от одного состояния структуры белка к другому прослеживается через матрицу расширения растущего выравнивания по одному фрагменту за раз. Оптимальный путь такой определяет комбинаторно-расширенное выравнивание. Веб-сервер, реализующий метод и предоставляющий базу данных парных сопоставлений структур в банке данных, расположен на веб-сайте Combinatorial Extension.
Филогенетика и выравнивание последовательностей - связанных области из общей необходимости оценки родства последовательностей. Область филогенетики широко использует выравнивание последовательностей при построении и интерпретации филогенетических, которые используются для эволюции отношений между гомологичными генами, представленными в геномы различных видов. Степень, в которой отслеживает в наборе запросы различаются, качественно с помощью эволюционных отрезков последовательностей друг от друга. Самого современного общего предка предполагает, что эта модель предполагает более древнюю. Это приближение, которое отражает гипотезу «молекулярные часы » о том, что есть слияния время), предполагает, что эффекты мутации и отбор постоянны во всех клонах последовательностей. Следовательно, он не учитывает возможные взаимодействия между организмами или варианты скоростей репарации ДНК или возможной функциональной консервации в последовательности. (В случае нуклеотидных последовательностей гипотез молекулярных часов в ее основной форме также учитывается разница в скорости принятия между молчащими мутациями, которые не изменяют значение данного кодона и другие мутации, которые приводят к в белок другой аминокислоты ). Более статистически точные методы точной организации эволюции на каждой ветви филогенетического дерева, тем самым более эффективные оценки времени слияния генов.
Методы прогрессивного множественного выравнивания по необходимости филогенетического дерева, потому что они включают в растущее выравнивание в родства. Другие методы, которые объединяют множественное выравнивание последовательностей и филогенетические деревья, сначала оценивают и сортируют деревья, а также вычисляют множественное выравнивание последовательностей из дерева с наивысшей оценкой. Обычно используемые методы построения филогенетического дерева в основном являются эвристическими, потому что проблема выбора оптимального дерева, как и проблема выбора оптимального множественного выравнивания последовательностей, является NP-сложной.
Выравнивание последовательностей полезно в биоинформатике для определения последовательностей, создания филогенетических деревьев и разработки моделей гомологии белковых структур. Однако биологическая значимость выравнивания последовательностей не всегда ясна. Частично, что совпадения отражают степень эволюционных изменений между последовательностями, происходящими общим от предка; однако формально, что конвергентная эволюция может вызвать очевидное сходство между белками, которые эволюционно не связаны, но имеют сходные функции и имеют сходные структуры.
При поиске в базе данных, таком как BLAST, статистические могут определять вероятность точно выравнивания методов между последовательностями или участками последовательностей, возникающих случайно, с учетом размера и состава базы, в котором выполняется поиск. Эти значения могут значительно различаться в зависимости от области поиска. В частности, вероятность случайного обнаружения данного выравнивания увеличивается, если база данных состоит только из последовательностей того же организма, что и запрашиваемая последовательность. Повторяющиеся последовательности в базе данных или запросе также могут исказить как результаты поиска, так и оценку статистической значимости; BLAST автоматически фильтрует такие повторяющиеся последовательности в запросе, чтобы избежать очевидных совпадений, которые являются статистическими артефактами.
Методы оценки статистической значимости выравниваний последовательностей с разрывом доступны в литературе.
Статистическая значимость указывает на вероятность того, что выравнивание данного качества может произойти случайно, но не указывает данное выравнивание превос альтернативное выравнивание тех же последовательностей. Меры достоверности выравнивания указывают на степень совмещения с наилучшей оценкой для данной последовательности по существу сходны. Методы оценки достоверности выравнивания для выравнивания последовательностей с разрывом доступны в литературе.
Выбор функции оценки, которая отражает биологические или статистические наблюдения за известными последовательностями, важен для достижения хорошего выравнивания. Последовательности белков часто выравниваются с использованием матриц замен, которые отражают вероятности заданных замен между символами. Серия матриц, называемая матрицей PAM (матрица точечной принятой мутации, используемая Маргарет Дейхофф и иногда называемая матрицами Дейхоффа ») явно кодирует эволюционные приближения, соответствующие скорости и вероятные исходные мутации. Другая распространенная серия матриц оценки, известная как BLOSUM (матрица за ущерб блоков), кодирует эмпирически данные вероятности за ущерб. Варианты обоих типов матриц используются для обнаружения последовательностей с разными уровнями дивергенции, что позволяет пользователям BLAST или FASTA ограничивать поиск более близкими совпадениями или расширять для обнаружения более расходящихся последовательностей. Штрафы за пробелы учитывают появление пробела - в эволюционной модели, вставки или удаления мутации - как в нуклеотидных, так и в белковых последовательностях, и поэтому значения штрафа должны быть пропорциональны ожидаемой последовательностью таких мутаций. Таким образом, качество произведенных выравниваний зависит от качества функции подсчета очков.
Может быть очень полезно и поучительно попробовать одно и то же выравнивание несколькими разными вариантами выбора для матрицы скоринга и / или значений штрафа за пробелы и сравнить результаты. Области, в решении которых является слабым или неуникальным, часто можно определить, какие области выравнивания устойчивы к изменениям выравнивания.
Секвенированная РНК, такая как теги экспрессированной последовательности и полноразмерные мРНК, могут быть сопоставлены с секвенированным геномом, чтобы найти, где находятся гены, и получить информацию об альтернативном соединении и. Выравнивание последовательностей также является частью сборки генома, где последовательности выравниваются для обнаружения перекрытия, чтобы можно было сформировать контиги (длинные участки последовательности). Другое применение - анализ SNP, при котором последовательности от разных индивидуумов выравниваются, чтобы найти отдельные пары оснований, которые часто различаются в популяции.
Используемые методы для выравнивания биологической последовательности также нашли применение в других областях, особенно в обработке естественного языка и в социальных науках, где алгоритм Нидлмана-Вунша обычно называют Оптимальным соответствие. Методы, которые генерируют набор элементов, из которых будут выбираться слова в алгоритмах генерации естественного языка, заимствовали множество методов выравнивания последовательностей из биоинформатики для создания лингвистических версий компьютерных математических доказательств. В области исторической и сравнительной лингвистики выравнивание последовательностей использовалось для частичной автоматизации сравнительный метод, с помощью которого лингвисты традиционно реконструируют языки. В бизнес-и маркетинговых исследованиях также применялись различные методы выравнивания последовательностей при анализе серий покупок с течением времени.
Более полный список доступного программного обеспечения с разбивкой по алгоритмам и типу выравнивания доступны на программное обеспечение для выравнивания последовательностей, но общие программные инструменты, используемые для общих задач выравнивания последовательностей, включая ClustalW2 и T-coffee для выравнивания, а также BLAST и FASTA3x для поиска в базе данных. Также доступны коммерческие инструменты, такие как DNASTAR Lasergene и PatternHunter. Инструменты, помеченные какющие выравнивание последовательностей, выполнение в реестре bio.tools.
Алгоритмы выравнивания и программное обеспечение можно напрямую сравнивать друг с другом, используя набор эталонных сравнительных сравнений последовательностей, известных как BAliBASE. Набор элементов состоит из структурных выравниваний, которые можно рассматривать как стандартные, с которыми сравниваются методы, основанные на последовательностях. Относительная эффективность многих распространенных методов при решении часто встречающихся проблем центров сведена в таблицу, а отдельные результаты опубликованы на сайте BAliBASE. Исчерпывающий список оценок BAliBASE для многих (в настоящее время 12) различных инструментов выравнивания может быть вычислен в рабочей среде для белков STRAP.
| Викиверситет имеет обучающие ресурсы по Точечной матрице методы |