
Войти
| Разработан | Wellcome Trust Sanger Institute |
|---|---|
| Первоначальный выпуск | ~ 2000 |
| Тип формата | Биоинформатика |
| Расширенный от | ASCII и Формат FASTA |
| Веб-сайт | maq.sourceforge.net / fastq.shtml |
Формат FASTQ - это текстовый формат для хранения как биологической последовательности (обычно нуклеотидной последовательности ), так и соответствующих показателей качества. И буква последовательности, и показатель качества для краткости кодируются одним символом ASCII.
Первоначально он был разработан в Wellcome Trust Sanger Institute для объединения последовательности в формате FASTA и данных о качестве, но недавно стал де-факто стандарт для хранения результатов высокопроизводительных инструментов секвенирования, таких как Illumina Genome Analyzer.
A FASTQ файл обычно использует четыре строки на последовательность.
Файл FASTQ, содержащий одну последовательность, может выглядеть следующим образом:
@SEQ_ID GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT +! '' * (((** * +)) %%% ++) (%%%%). 1 *** - + * '')) ** 55CCF>>>>>>1970C65
Байт, представляющий качество, начинается с От 0x21 (низкое качество; '!' В ASCII) до 0x7e (высшее качество; '~' в ASCII). Вот символы значения качества в порядке возрастания качества слева направо (ASCII ):
! "# $% '() * +, -. / 0123456789 :; <=>? @ABCDEFGHIJKLMNOPQRSTUVWXYZ [\] ^ _ ʻabcdefghijklmnopqrstuvwxyz {|} ~Исходные файлы Sanger FASTQ также позволяли обертывать строки последовательности и качества (разбивать на несколько строк), но это обычно не рекомендуется поскольку это может усложнить синтаксический анализ из-за неудачного выбора "@" и "+" в качестве маркеров (эти символы также могут встречаться в строке качества).
Последовательности из программное обеспечение Illumina использует систематический идентификатор:
@ HWUSI-EAS100R: 6: 73: 941: 1973 # 0/1
| HWUSI-EAS100R | уникальный имя прибора |
|---|---|
| 6 | дорожка проточной кюветы |
| 73 | номер плитки в полосе проточной кюветы |
| 941 | 'x' -координата кластера внутри плитки |
| 1973 | 'y'-координата кластера в тайле |
| # 0 | номер индекса для мультиплексированной выборки (0 для no i ndexing) |
| / 1 | член пары, / 1 или / 2 (парный конец или сопряженная пара только для чтения) |
Версии конвейера Illumina, начиная с 1.4, похоже, используют #NNNNNN вместо # 0 для идентификатора мультиплекса, где NNNNNN - это последовательность тега мультиплексирования.
В Casava 1.8 формат строки '@' изменился:
@ EAS139: 136: FC706VJ: 2: 2104: 15343: 197393 1: Y: 18: ATCACG
| EAS139 | уникальное имя прибора |
|---|---|
| 136 | идентификатор прогона |
| FC706VJ | идентификатор проточной ячейки |
| 2 | полоса проточной ячейки |
| 2104 | номер тайла в полосе проточной ячейки |
| 15343 | 'x' координата кластера внутри тайла |
| 197393 | 'y' координата кластера в тайле |
| 1 | член пары, 1 или 2 (только чтение парных или сопряженных пар) |
| Y | Y, если чтение отфильтровано (не прошло), N в противном случае |
| 18 | 0, когда ни один из управляющих битов не включен, в противном случае это четное число |
| ATCACG | последовательность индексов |
Обратите внимание, что более поздние версии программного обеспечения Illumina выводят номер образца (взятый из таблицы образцов) вместо индексной последовательности. Например, следующий заголовок может появиться в первом примере пакета:
@ EAS139: 136: FC706VJ: 2: 2104: 15343: 197393 1: N: 18: 1
Файлы FASTQ из архива INSDC чтения последовательности часто включают описание, например
@ SRR001666.1 071112_SLXA-EAS1_s_7: 5: 1: 817: 345 длина = 36 GGGTGATGGCCGCTGCCGATGGCGTCAAATCCCACC + SRR001666.1 071112_SLXA-EAS1_s_7ICIIIIIIIIII5: длина 1: 8BIGIIIIII: 1 В этом примере есть идентификатор, присвоенный NCBI, а описание содержит исходный идентификатор из Solexa / Illumina (как описано выше) плюс длину чтения. Секвенирование выполняли в режиме парных концов (размер вставки ~ 500 пар оснований), см. SRR001666. Формат вывода по умолчанию fastq-dump создает целые пятна, содержащие любые технические чтения и, как правило, одно- или парные биологические чтения.$ fastq-dump.2.9.0 -Z -X 2 SRR001666 Чтение 2 точек для SRR001666 Записано 2 точек для SRR001666 @ SRR001666.1 071112_SLXA-EAS1_s_7: 5: 1: 817: 345 длина = 72 GGGTGATGATCATCAAGTAGTAGTAGTAGTACTACTAGTAGTAGTAGTACCACTAGTAGTAGTAGTACCACTGACTAGTAGTACCAGTAGTACCAGTAGTAG 071112_SLXA-EAS1_s_7: 5: 1: 817: 345 длина = 72 IIIIIIIIIIIIIIIIIIIIIIIIIIIIII9IG9ICIIIIIIIIIIIIIIIIIIIIDIIIIIII>IIIIII / @ SRR001666.2 071112_SLXA-EAS1_s_7: 5: 1: 801: 338 длина = 72 GTTCAGGGATACGACGTTTGTATTTTAAGAATCTGAAGCAGAAGTCGATGATAATACGCGTCGTTTTATCAT + SRR001666.2 071112_SLXA-EAS1_s_7: 5: 1: 801 : 338 length = 72 IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII6IBIIIIIIIIIIIIIIIIIIIGII>IIIII-I) 8IСовременное использование FASTQ почти всегда включает разбиение пятна на его биологические чтения, как описано в метаданных, предоставленных отправителем:
$ fastq-dump -X - 2 SRR001666 -split-3 Читать 2 точки для SRR001666 Написано 2 точки для SRR001666 $ head SRR001666_1.fastq SRR001666_2.fastq ==>SRR001666_1.fastq <== @SRR001666.1 071112_SLXA-EAS1_s_7:5:1:817:345 length=36 GGGTGATGGCCGCTGCCGATGGCGTCAAATCCCACC +SRR001666.1 071112_SLXA-EAS1_s_7:5:1:817:345 length=36 IIIIIIIIIIIIIIIIIIIIIIIIIIIIII9IG9IC @SRR001666.2 071112_SLXA-EAS1_s_7:5:1:801:338 length=36 GTTCAGGGATACGACGTTTGTATTTTAAGAATCTGA +SRR001666.2 071112_SLXA-EAS1_s_7:5:1:801:338 length=36 IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII6IBI ==>SRR001666_2.fastq <== @SRR001666.1 071112_SLXA-EAS1_s_7:5:1:817:345 length=36 AAGTTACCCTTAACAACTTAAGGGTTTTCAAATAGA +SRR001666.1 071112_SLXA-EAS1_s_7:5:1:817:345 length=36 IIIIIIIIIIIIIIIIIIIIDIIIIIII>IIIIII / @ SRR001666.2 071112_SLXA_7: 51112_SLXA-EAS1: 5 : 801: 338 length = 36 AGCAGAAGTCGATGATAATACGCGTCGTTTTATCAT + SRR001666.2 071112_SLXA-EAS1_s_7: 5: 1: 801: 338 length = 36 IIIIIIIIIIIIIIIIIIIIIIGII>IIIII-I) 8IПри чтении в исходном формате можно попытаться быстро восстановить дамп. NCBI не хранит исходные имена чтения по умолчанию:
$ fastq-dump -X 2 SRR001666 --split-3 --origfmt Прочитать 2 точки для SRR001666 Написано 2 точки для SRR001666 $ head SRR001666_1.fastq SRR001666_2.fastq ==>SRR001666_1.fastq <== @071112_SLXA-EAS1_s_7:5:1:817:345 GGGTGATGGCCGCTGCCGATGGCGTCAAATCCCACC +071112_SLXA-EAS1_s_7:5:1:817:345 IIIIIIIIIIIIIIIIIIIIIIIIIIIIII9IG9IC @071112_SLXA-EAS1_s_7:5:1:801:338 GTTCAGGGATACGACGTTTGTATTTTAAGAATCTGA +071112_SLXA-EAS1_s_7:5:1:801:338 IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII6IBI ==>SRR001666_2.fastq <== @071112_SLXA-EAS1_s_7:5:1:817:345 AAGTTACCCTTAACAACTTAAGGGTTTTCAAATAGA +071112_SLXA-EAS1_s_7:5:1:817:345 IIIIIIIIIIIIIIIIIIIIDIIIIIII>IIIIII / @ 071112_SLXA-EAS1_s_7: 5: 1: 801: 338 AGCAGAAGTCGATGATAATACGCGTCGTTTTATCAT + 071112_SLXA-EAS1_s_7: 5: 1: 801: 338 IIIIIIIIIIIIIIIIIIIIIIGII>IIIIII) 8ИВ В приведенном выше примере использовались исходные имена чтения, а не присоединенное имя чтения. Запуски доступа NCBI и содержащиеся в них чтения. Исходные имена чтения, присвоенные секвенсорами, могут функционировать как локальные уникальные идентификаторы чтения и передавать столько же информации, сколько и серийный номер. Приведенные выше идентификаторы были присвоены алгоритмически на основе информации о прогоне и геометрических координат. Ранние загрузчики SRA анализировали эти идентификаторы и хранили свои разложенные компоненты внутри. NCBI прекратил записывать прочитанные имена, потому что они часто изменяются по сравнению с исходным форматом поставщиков, чтобы связать некоторую дополнительную информацию, значимую для конкретного конвейера обработки, и это вызвало нарушения формата имени, что привело к большому количеству отклоненных представлений. Без четкой схемы для имен чтения их функция остается функцией уникального идентификатора чтения, передавая тот же объем информации, что и серийный номер чтения. См. Различные проблемы с инструментарием SRA для получения подробной информации и обсуждения.
Также обратите внимание, что fastq-dump преобразует эти данные FASTQ из исходной кодировки Solexa / Illumina в стандарт Sanger (см. Кодировки ниже). Это связано с тем, что SRA служит хранилищем информации NGS, а не форматом. Различные инструменты * -dump могут создавать данные в нескольких форматах из одного источника. Требования к этому были продиктованы пользователями в течение нескольких лет, при этом большая часть раннего спроса исходила от 1000 Genomes Project.
VariationsQuality
Значение качества Q представляет собой целочисленное отображение p (т. е. вероятность того, что соответствующий базовый вызов неверен). Использовались два разных уравнения. Первый - это стандартный вариант Сэнгера для оценки надежности базового вызова, иначе известный как показатель качества Phred :
Конвейер Solexa (т. е. программное обеспечение, поставляемое с анализатором генома Illumina) ранее использовал другое сопоставление, кодируя шансы p / (1-p) вместо вероятности p:
Хотя оба сопоставления асимптотически идентичны при более высоких значениях качества, они различаются на более низких уровнях качества (т. е. приблизительно p>0,05 или, что эквивалентно, Q < 13).
Взаимосвязь между Q и p с использованием уравнений Сенгера (красный) и Solexa (черный) (описанных выше). Вертикальная пунктирная линия указывает на p = 0,05 или, что эквивалентно, Q ≈ 13.
Иногда возникали разногласия. о том, какое отображение на самом деле использует Illumina. Руководство пользователя (Приложение B, стр. 122) для версии в 1.4 конвейера Illumina говорится, что: «Оценка определяется как Q = 10 * log10 (p / (1-p)) [sic ], где p - вероятность базового вызова, соответствующего рассматриваемая база ". Оглядываясь назад, кажется, что эта запись в руководстве была ошибкой. В руководстве пользователя (Что нового, стр. 5) для версии 1.5 конвейера Illumina вместо этого приводится следующее описание: «Важные изменения в конвейере v1.3 [sic ]. Схема оценки качества была изменена на Phred [ то есть, Sanger] схема оценки, закодированная как символ ASCII путем добавления 64 к значению Phred. Оценка Phred по основанию:
, где e - оценка вероятности неправильного основания.
Кодирование
@ HWI-EAS209_0006_FC706VJ: 5: 58: 5894: 21141 # ATCACG / 1 TTAATTGGTAAATAAATCTCCTAATAGCTTAGATNTTACCTTNNNNNNNNNNTAGTTTCTTGAGATTTGTTGGGGGAGACATTTTTGTGATTGCCTTGAT + HWI-EAS209_0006_FC706VJ: 5: 58: 5894: 21141 # ATCACG / 1 efcfffffcfeefffcffffffddf`feed] `] _Ba _ ^ __ [YBBBBBBBBBBRTT \]] dddd`ddd ^ dddadd ^ BBBBBBBBBBBBBBBBBBBBBBBB
альтернативная интерпретация этого Было предложено кодирование ASCII. Кроме того, в запусках Illumina с использованием элементов управления PhiX символ «B» представлял «неизвестный показатель качества». Уровень ошибок при чтении «B» был примерно на 3 балла по шкале phred ниже среднего наблюдаемого балла для данного прогона.
Для необработанных чтений диапазон оценок будет зависеть от технологии и используемого основного вызывающего объекта, но обычно будет до 41 для недавней химии Illumina. Поскольку ранее максимальная наблюдаемая оценка качества составляла всего 40, различные скрипты и инструменты ломаются, когда они сталкиваются с данными со значениями качества, превышающими 40. Для обработанных чтений оценки могут быть даже выше. Например, значения качества 45 наблюдаются при чтениях из службы последовательного считывания Illumina (ранее Moleculo).
SSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSS............................................................................... XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX..................................................... IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII....................................................... Дж JJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJ..................... LLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLL.................................................... PPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPPP7 ? @ABCDEFGHIJKLMNOPQRSTUVWXYZ [\] ^ _ ʻabcdefghijklmnopqrstuvwxyz {|} ~ | | | | | 33 59 64 73 104 126 0........................26... 31....... 40-5.... 0........ 9............................. 40 0 ........ 9............................. 40 3..... 9.............................. 41 0,2...................... 26... 31........ 41 0.................. 20........ 30........ 40........ 50.......................................... 93S - Sanger Phred + 33, обычное чтение обычно (0, 40) X - Solexa Solexa +64, необработанные чтения обычно (-5, 40) I - Illumina 1.3+ Phred + 64, необработанные чтения обычно (0, 40) J - Illumina 1.5+ Phred + 64, необработанные чтения обычно (3, 41) с 0 = неиспользуемые, 1 = не используется, 2 = индикатор контроля качества сегмента чтения (жирный шрифт) (Примечание: см. Обсуждение выше). L - Illumina 1.8+ Phred + 33, обычное чтение обычно (0, 41) P - PacBio Phred + 33, HiFi обычно читает (0, 93)
Для данных SOLiD, последовательность находится в цветовом пространстве, кроме первой позиции. Значения качества соответствуют формату Sanger. Инструменты выравнивания различаются по своей предпочтительной версии значений качества: некоторые включают оценку качества (установленную на 0, т. Е. '!') Для ведущего нуклеотида, другие - нет. Архив чтения последовательности включает этот показатель качества.
Моделирование чтения FASTQ реализовано несколькими инструментами. Сравнение этих инструментов можно увидеть здесь.
Универсальные инструменты, такие как Gzip и bzip2, рассматривают FASTQ как простой текстовый файл и в результате неоптимальные степени сжатия. Архив чтения последовательности NCBI кодирует метаданные, используя схему LZ-77. Общие компрессоры FASTQ обычно сжимают отдельные поля (считанные имена, последовательности, комментарии и оценки качества) в файле FASTQ отдельно; к ним относятся DSRC и DSRC2, FQC, LFQC, Fqzcomp и Slimfastq.
Удобно иметь эталонный геном, потому что тогда вместо хранения самих нуклеотидных последовательностей можно просто выровнять чтения с эталонным геномом и сохранить позиции (указатели) и несовпадения; указатели затем могут быть отсортированы в соответствии с их порядком в эталонной последовательности и закодированы, например, с кодированием длин серий. Когда покрытие или содержание повторов в секвенированном геноме высокое, это приводит к высокой степени сжатия. В отличие от форматов SAM / BAM, файлы FASTQ не определяют эталонный геном. Компрессоры FASTQ на основе выравнивания поддерживают использование либо предоставленных пользователем, либо созданных de novo ссылок: LW-FQZip использует предоставленный эталонный геном, а Quip, Leon, k-Path и KIC выполняют de novo с использованием подхода на основе графа де Брейна.
Явное отображение чтения и сборка de novo обычно выполняются медленно. Компрессоры FASTQ на основе переупорядочения сначала кластер читает, что разделяет длинные подстроки, а затем независимо сжимает чтения в каждом кластере после их переупорядочения или объединения в более длинные контиги, достигая, возможно, наилучшего компромисса между время работы и степень сжатия. SCALCE - первый такой инструмент, за ним следуют Orcom и Mince. BEETL использует обобщенное преобразование Барроуза – Уиллера для переупорядочения операций чтения, а HARC обеспечивает лучшую производительность за счет переупорядочения на основе хешей. AssemblTrie вместо этого собирает операции чтения в деревья ссылок с минимальным общим количеством символов в ссылке.
Тесты для этих инструментов доступны в.
Значения качества приходится около половины необходимого дискового пространства в формате FASTQ (до сжатия), поэтому сжатие значений качества может значительно снизить требования к хранению и ускорить анализ и передачу данных секвенирования. В последнее время в литературе рассматриваются как сжатие без потерь, так и сжатие с потерями. Например, алгоритм QualComp выполняет сжатие с потерями со скоростью (количество бит на значение качества), указанной пользователем. Основываясь на результатах теории искажения скорости, он распределяет количество битов так, чтобы минимизировать MSE (среднеквадратичную ошибку) между исходным (несжатым) и восстановленным (после сжатия) значениями качества. Другие алгоритмы сжатия значений качества включают SCALCE и Fastqz. Оба являются алгоритмами сжатия без потерь, которые обеспечивают дополнительный подход к управляемому преобразованию с потерями. Например, SCALCE уменьшает размер алфавита на основании наблюдения, что «соседние» значения качества в целом похожи. Для эталонного теста см.
Начиная с HiSeq 2500 Illumina дает возможность выводить качество, которое было грубо измельчено, в ячейки качества. Разделенные оценки вычисляются непосредственно из таблицы эмпирических показателей качества, которая сама привязана к оборудованию, программному обеспечению и химическим характеристикам, которые использовались во время эксперимента по секвенированию.
Шифрование файлов FASTQ в основном решалась с помощью специального инструмента шифрования: Cryfa. Cryfa использует шифрование AES и позволяет уплотнять данные помимо шифрования. Он также может обращаться к файлам FASTA.
Не существует стандартного расширения для файла FASTQ, но обычно используются.fq и.fastq.


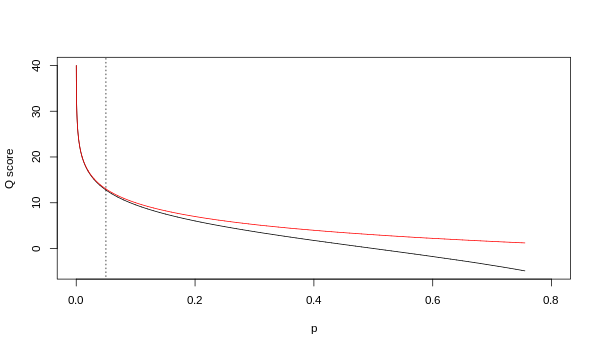 Взаимосвязь между Q и p с использованием уравнений Сенгера (красный) и Solexa (черный) (описанных выше). Вертикальная пунктирная линия указывает на p = 0,05 или, что эквивалентно, Q ≈ 13.
Взаимосвязь между Q и p с использованием уравнений Сенгера (красный) и Solexa (черный) (описанных выше). Вертикальная пунктирная линия указывает на p = 0,05 или, что эквивалентно, Q ≈ 13.