
Войти
 Сводка RNA-Seq. Внутри организма гены транскрибируются и (в эукариотическом организме ) сплайсируются для получения зрелых транскриптов мРНК (красный). МРНК извлекается из организма, фрагментируется и копируется в стабильную дц-кДНК (синий). Дц-кДНК секвенируют с использованием высокопроизводительных методов секвенирования с коротким считыванием. Затем эти последовательности могут быть выровнены с эталонной последовательностью генома, чтобы реконструировать, какие участки генома транскрибируются. Эти данные можно использовать для аннотации того, где находятся экспрессируемые гены, их относительных уровней экспрессии и любых альтернативных вариантов сплайсинга.
Сводка RNA-Seq. Внутри организма гены транскрибируются и (в эукариотическом организме ) сплайсируются для получения зрелых транскриптов мРНК (красный). МРНК извлекается из организма, фрагментируется и копируется в стабильную дц-кДНК (синий). Дц-кДНК секвенируют с использованием высокопроизводительных методов секвенирования с коротким считыванием. Затем эти последовательности могут быть выровнены с эталонной последовательностью генома, чтобы реконструировать, какие участки генома транскрибируются. Эти данные можно использовать для аннотации того, где находятся экспрессируемые гены, их относительных уровней экспрессии и любых альтернативных вариантов сплайсинга. RNA-Seq (названный аббревиатурой от «секвенирования РНК») - это особая технология, основанная на секвенирование метод, который использует секвенирование следующего поколения (NGS) для выявления присутствия и количества РНК в биологическом образце в данный момент, анализируя непрерывно изменяющиеся клеточные транскриптом.
В частности, RNA-Seq облегчает возможность просмотра транскриптов сращивания альтернативных генов, посттранскрипционных модификаций, слияния генов, мутаций / SNP и изменения в экспрессии генов с течением времени или различия в экспрессии генов в разных группах или в разных обработках. Помимо транскриптов мРНК, RNA-Seq может анализировать различные популяции РНК, включая общую РНК, малую РНК, такую как miRNA, тРНК и профилирование рибосом. RNA-Seq также может использоваться для определения границ экзона / интрона и проверки или изменения ранее аннотированных границ гена 5' и 3 '. Последние достижения в области RNA-Seq включают секвенирование отдельных клеток и секвенирование фиксированной ткани in situ.
До RNA-Seq исследования экспрессии генов проводились с использованием микрочипов на основе гибридизации. Проблемы с микрочипами включают артефакты перекрестной гибридизации, плохую количественную оценку низко и высоко экспрессируемых генов и необходимость знать последовательность априори. Из-за этих технических проблем транскриптомика перешла на методы, основанные на секвенировании. Они перешли от секвенирования по Сэнгеру из библиотек экспрессируемых последовательностей к методам на основе химических меток (например, серийный анализ экспрессии генов ) и, наконец, к текущим технология, секвенирование следующего поколения кДНК (в частности, RNA-Seq).
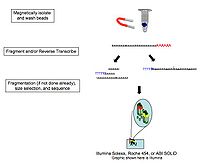
 Обзор типичного RNA-Seq экспериментальный рабочий процесс.
Обзор типичного RNA-Seq экспериментальный рабочий процесс. Общие шаги по подготовке библиотеки комплементарной ДНК (кДНК) для секвенирования описаны ниже, но часто варьируются в зависимости от платформы.
| Стратегия | Тип РНК | Содержание рибосомной РНК | Содержание необработанной РНК | Содержание геномной ДНК | Метод выделения |
|---|---|---|---|---|---|
| Общая РНК | Все | Высокий | Высокий | Высокий | Нет |
| Выбор PolyA | Кодирование | Низкое | Низкое | Низкое | Гибридизация с поли (dT) олигомерами |
| истощение рРНК | Кодирование, некодирование | Низкое | Высокая | Высокая | Удаление олигомеров, комплементарных рРНК |
| Захват РНК | Нацеленная | Низкая | Умеренная | Низкая | Гибридизация с зондами, комплементарными желаемым транскриптам |
При секвенировании РНК, отличной от мРНК, подготовка библиотеки изменяется. Клеточная РНК выбирается на основе желаемого диапазона размеров. Для малых РНК-мишеней, таких как miRNA, РНК выделяют путем отбора по размеру. Это может быть выполнено с помощью геля для исключения размера, с помощью магнитных шариков для выбора размера или с помощью коммерчески разработанного набора. После выделения линкеры добавляют к 3'- и 5'-концам, а затем очищают. Последним этапом является создание кДНК посредством обратной транскрипции.

Поскольку преобразование РНК в кДНК, лигирование, амплификация и другие манипуляции с образцами, как было показано, вносят систематические ошибки и артефакты, которые могут мешать как правильной характеризации, так и количественной оценке транскриптов, прямое секвенирование РНК одной молекулы было исследовано компаниями, включая Helicos (банкрот), Oxford Nanopore Technologies и другими. Эта технология обеспечивает прямую последовательность молекул РНК массово-параллельным образом.
Стандартные методы, такие как микроматрицы и стандартный массовый анализ RNA-Seq, анализируют экспрессию РНК из больших популяций клеток. В смешанных популяциях клеток эти измерения могут скрыть важные различия между отдельными клетками в этих популяциях.
Секвенирование одноклеточной РНК (scRNA-Seq) обеспечивает профили экспрессии отдельных клеток. Хотя невозможно получить полную информацию о каждой РНК, экспрессируемой каждой клеткой, из-за небольшого количества доступного материала, образцы экспрессии генов могут быть идентифицированы с помощью анализа кластеризации генов . Это может раскрыть существование в популяции клеток редких типов клеток, которые, возможно, никогда раньше не наблюдались. Например, редкие специализированные клетки в легких, называемые легочными ионоцитами, которые экспрессируют регулятор трансмембранной проводимости при муковисцидозе, были идентифицированы в 2018 году двумя группами, выполнявшими scRNA-Seq на эпителии дыхательных путей легких.
 Рабочий процесс секвенирования одноклеточной РНК
Рабочий процесс секвенирования одноклеточной РНК Текущие протоколы scRNA-Seq включают следующие этапы: выделение отдельной клетки и РНК, обратная транскрипция (RT), амплификация, библиотека генерация и секвенирование. Ранние методы разделяли отдельные клетки на отдельные лунки; более современные методы инкапсулируют отдельные клетки в капельки в микрофлюидном устройстве, где происходит реакция обратной транскрипции, превращающая РНК в кДНК. Каждая капля несет на себе «штрих-код» ДНК, который однозначно маркирует кДНК, полученные из одной клетки. После завершения обратной транскрипции кДНК из многих клеток могут быть смешаны вместе для секвенирования; транскрипты из конкретной клетки идентифицируются уникальным штрих-кодом.
Проблемы с scRNA-Seq включают сохранение первоначального относительного количества мРНК в клетке и идентификацию редких транскриптов. Этап обратной транскрипции имеет решающее значение, поскольку эффективность реакции RT определяет, какая часть популяции РНК клетки будет в конечном итоге проанализирована секвенатором. Процессивность обратных транскриптаз и используемые стратегии прайминга могут влиять на продукцию полноразмерной кДНК и создание библиотек, смещенных в сторону 3 ’или 5’ конца генов.
На стадии амплификации для амплификации кДНК в настоящее время используется либо ПЦР, либо in vitro транскрипция (IVT). Одним из преимуществ методов на основе ПЦР является возможность генерировать полноразмерную кДНК. Однако различная эффективность ПЦР для конкретных последовательностей (например, содержимого GC и структуры snapback) также может быть экспоненциально усилена, создавая библиотеки с неравномерным покрытием. С другой стороны, хотя библиотеки, созданные с помощью IVT, могут избежать смещения последовательности, вызванного ПЦР, определенные последовательности могут транскрибироваться неэффективно, что вызывает выпадение последовательности или генерирование неполных последовательностей. Было опубликовано несколько протоколов scRNA-Seq: Tang et al., STRT, SMART-seq, CEL-seq, RAGE-seq, Quartz-seq. и C1-CAGE. Эти протоколы различаются с точки зрения стратегий обратной транскрипции, синтеза и амплификации кДНК, а также возможности размещения штрих-кодов, специфичных для последовательности (например, UMI ), или способности обрабатывать объединенные образцы.
В 2017 г. были представлены два подхода для одновременного измерения экспрессии мРНК и белка в отдельных клетках с помощью олигонуклеотидно-меченных антител, известных как REAP-seq, и CITE-seq.
scRNA-Seq становится широко используется в биологических дисциплинах, включая развитие, неврологию, онкологию, аутоиммунное заболевание и инфекционное заболевание.
. scRNA-Seq предоставил значительное понимание развитие эмбрионов и организмов, в том числе червя Caenorhabditis elegans и регенеративного планария. Первыми позвоночными животными, которые были нанесены на карту таким образом, были данио и Xenopus laevis. В каждом случае изучались несколько стадий эмбриона, что позволяло картировать весь процесс развития для каждой клетки. Наука признала эти достижения как «прорыв года» в 2018 году .
При разработке и проведении экспериментов с RNA-Seq учитывается множество параметров :
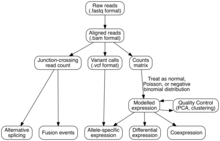 Схема, описывающая анализ RNA-Seq, описанный в этом разделе
Схема, описывающая анализ RNA-Seq, описанный в этом разделе Два метода используются для присвоения необработанных считываний последовательности геномным признакам (т. е. сборка транскриптома):
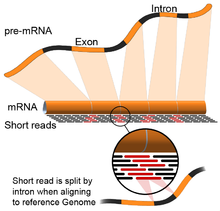 картирование RNA-Seq коротких прочтений в соединениях экзон-экзон. Конечная мРНК секвенируется, в ней отсутствуют интронные участки пре-мРНК.
картирование RNA-Seq коротких прочтений в соединениях экзон-экзон. Конечная мРНК секвенируется, в ней отсутствуют интронные участки пре-мРНК. Примечание о качестве сборки: в настоящее время консенсус заключается в том, что 1) качество сборки может варьироваться в зависимости от используемой метрики, 2) сборки, которые хорошо оцененные у одного вида не обязательно хорошо проявляют себя у другого вида, и 3) сочетание различных подходов может быть наиболее надежным.
Экспрессия оценивается количественно для изучения клеточных изменений в реакция на внешние раздражители, различия между здоровыми и больными состояниями и другие вопросы исследования. Экспрессия генов часто используется в качестве показателя изобилия белка, но они часто не эквивалентны из-за посттранскрипционных событий, таких как РНК-интерференция и нонсенс-опосредованный распад.
Экспрессия количественно оценивается путем подсчета количество чтений, которые сопоставлены с каждым локусом на этапе сборки транскриптома. Экспрессию экзонов или генов можно количественно оценить с помощью контигов или аннотаций эталонных транскриптов. Эти наблюдаемые подсчеты считывания RNA-Seq были тщательно проверены на соответствие более старым технологиям, включая экспрессионные микроматрицы и qPCR. Примерами инструментов для количественной оценки количества являются HTSeq, FeatureCounts, Rcount, maxcounts, FIXSEQ и Cuffquant. Счетчики чтения затем преобразуются в соответствующие показатели для проверки гипотез, регрессии и других анализов. Параметры для этого преобразования:
Абсолютная количественная оценка экспрессии гена невозможна в большинстве экспериментов с RNA-Seq, которые определяют количественную экспрессию относительно всех транскриптов. Это возможно при выполнении RNA-Seq со вставками, образцов РНК в известных концентрациях. После секвенирования подсчет считываний всплесков последовательностей используется для определения взаимосвязи между счетчиками считывания каждого гена и абсолютными количествами биологических фрагментов. В одном примере этот метод использовался у эмбрионов Xenopus tropicalis для определения кинетики транскрипции.
Самым простым, но часто наиболее эффективным использованием RNA-Seq является обнаружение различия в экспрессии генов между двумя или более состояниями (например, лечили и не лечили); этот процесс называется дифференциальным выражением. Выходы часто называют дифференциально экспрессируемыми генами (DEG), и эти гены могут регулироваться либо с повышением, либо с понижением (т.е. повышать или понижать в интересующем состоянии). Есть много инструментов , которые выполняют дифференциальное выражение. Большинство из них выполняется в R, Python или в командной строке Unix. Обычно используемые инструменты включают DESeq, edgeR и voom + limma, все из которых доступны через R / Bioconductor. Вот общие соображения при выполнении дифференциальной экспрессии:
Последующий анализ для списка дифференциально экспрессируемых генов бывает двух видов, подтверждающих наблюдения и делая биологические выводы. Из-за ошибок дифференциальной экспрессии и RNA-Seq важные наблюдения воспроизводятся (1) ортогональным методом в тех же образцах (например, ПЦР в реальном времени ) или (2) другим, иногда предварительно зарегистрированный, эксперимент в новой когорте. Последнее помогает обеспечить обобщаемость и, как правило, может сопровождаться метаанализом всех объединенных когорт. Наиболее распространенный метод получения более высокого уровня биологического понимания результатов - это анализ обогащения набора генов, хотя иногда используются подходы к генам-кандидатам. Обогащение набора генов определяет, является ли перекрытие между двумя наборами генов статистически значимым, в этом случае перекрытие между дифференциально экспрессируемыми генами и наборами генов из известных путей / баз данных (например, Gene Ontology, KEGG, Онтология фенотипа человека ) или из дополнительных анализов тех же данных (например, сетей коэкспрессии). Общие инструменты для обогащения набора генов включают веб-интерфейсы (например, ENRICHR, g: profiler) и пакеты программного обеспечения. При оценке результатов обогащения одна эвристика состоит в том, чтобы сначала искать обогащение известной биологии как проверку работоспособности, а затем расширять область поиска для поиска новой биологии.
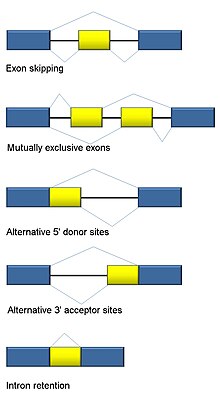 Альтернативные типы событий сплайсинга РНК. Экзоны представлены в виде синих и желтых блоков, интроны - в виде линий между ними.
Альтернативные типы событий сплайсинга РНК. Экзоны представлены в виде синих и желтых блоков, интроны - в виде линий между ними. Сплайсинг РНК является неотъемлемой частью эукариот и вносит значительный вклад в регуляцию и разнообразие белков, встречающийся в>90% генов человека. Существует несколько альтернативных режимов сплайсинга : пропуск экзонов (наиболее распространенный режим сплайсинга у людей и высших эукариот), взаимоисключающие экзоны, альтернативные донорные или акцепторные сайты, удержание интронов (наиболее распространенный режим сплайсинга у растений, грибов и т. простейшие), альтернативный сайт начала транскрипции (промотор) и альтернативное полиаденилирование. Одна из целей RNA-Seq - выявить альтернативные события сплайсинга и проверить, различаются ли они в разных условиях. При долгосрочном секвенировании фиксируется полный транскрипт и, таким образом, сводятся к минимуму многие проблемы при оценке распространенности изоформ, такие как отображение неоднозначного чтения. Для короткого чтения RNA-Seq существует несколько методов обнаружения альтернативного сплайсинга, которые можно разделить на три основные группы:
Инструменты дифференциальной экспрессии генов также могут использоваться для дифференциальной экспрессии изоформ, если изоформы предварительно количественно определены с помощью других инструментов, таких как RSEM.
Сети коэкспрессии представляют собой полученные из данных представления генов, ведущих себя одинаково в разных тканях и экспериментальных условиях. Их основная цель заключается в генерировании гипотез и подходах по принципу «чувство вины по ассоциации» для определения функций ранее неизвестных генов. Данные RNA-Seq были использованы для вывода генов, участвующих в определенных путях, на основе корреляции Пирсона как у растений, так и у млекопитающих. Основное преимущество данных RNA-Seq в этом виде анализа по сравнению с платформами микрочипов - это способность покрывать весь транскриптом, что позволяет получить более полные представления о сетях регуляции генов. Дифференциальная регуляция изоформ сплайсинга одного и того же гена может быть обнаружена и использована для прогнозирования их биологических функций. Взвешенный сетевой анализ коэкспрессии генов успешно использовался для идентификации модулей коэкспрессии и генов внутримодульных концентраторов на основе по данным последовательности РНК. Модули коэкспрессии могут соответствовать типам клеток или путям. Внутримодульные концентраторы с высокой степенью связи могут быть интерпретированы как представители соответствующего модуля. Собственный ген - это взвешенная сумма экспрессии всех генов в модуле. Собственные гены - полезные биомаркеры (признаки) для диагностики и прогноза. Были предложены подходы к трансформации, стабилизирующей отклонения, для оценки коэффициентов корреляции на основе данных seq РНК.
RNA-Seq фиксирует вариации ДНК, включая однонуклеотидные варианты, небольшие вставки / удаления. и структурная вариация. Вызов варианта в RNA-Seq аналогичен вызову варианта ДНК и часто использует те же инструменты (включая SAMtools mpileup и GATK HaplotypeCaller) с настройками для учета сплайсинга. Одним из уникальных параметров для вариантов РНК является аллель-специфическая экспрессия (ASE) : варианты только одного гаплотипа могут предпочтительно экспрессироваться из-за регуляторных эффектов, включая импринтинг и количественный признак экспрессии. локусы и некодирующие редкие варианты. Ограничения идентификации варианта РНК включают то, что он отражает только экспрессируемые области (у людей <5% of the genome) and has lower quality when compared to direct DNA sequencing.
Наличие совпадающих геномных и транскриптомных последовательностей человека может помочь в обнаружении посттранскрипционных изменений. -transcriptional edits (). Событие посттранскрипционной модификации идентифицируется, если транскрипт гена имеет аллель / вариант, не наблюдаемый в геномных данных.
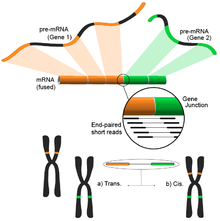 Картирование коротких считываний RNA-Seq по соединениям экзон-экзон, в зависимости от того, где каждый Конечные карты, это может быть определено как событие Trans или Cis.
Картирование коротких считываний RNA-Seq по соединениям экзон-экзон, в зависимости от того, где каждый Конечные карты, это может быть определено как событие Trans или Cis. Вызванные различными структурными модификациями в геноме гены слияния привлекли внимание из-за их связи с раком. Способность RNA-Seq для беспристрастного анализа всего транскриптома образца делает его привлекательным инструментом для поиска таких общих событий при раке.
Идея вытекает из процесса согласования коротких транскриптомных чтений с повторным геном ференции. Большинство коротких прочтений попадают в один полный экзон, и ожидается, что меньший, но все же большой набор будет отображаться на известные экзон-экзонные соединения. Оставшиеся неотмеченные короткие считывания затем будут дополнительно проанализированы, чтобы определить, соответствуют ли они соединению экзон-экзон, где экзоны происходят от разных генов. Это свидетельствовало бы о возможном событии слияния, однако из-за продолжительности считывания это могло оказаться очень шумным. Альтернативный подход заключается в использовании считывания на конце пары, когда потенциально большое количество парных считываний будет отображать каждый конец на другой экзон, обеспечивая лучшее покрытие этих событий (см. Рисунок). Тем не менее, конечный результат состоит из множества и потенциально новых комбинаций генов, обеспечивающих идеальную отправную точку для дальнейшей проверки.
 Количество рукописей на PubMed, содержащих RNA-Seq, все еще увеличивается.
Количество рукописей на PubMed, содержащих RNA-Seq, все еще увеличивается. RNA-Seq была впервые разработана в середине 2000-х годов с появлением секвенирования следующего поколения технологии. Первые рукописи, которые использовали RNA-Seq даже без использования термина, включают рукописи рака простаты клеточных линий (датированы 2006 г.), Medicago truncatula (2006), кукуруза (2007) и Arabidopsis thaliana (2007), а сам термин «RNA-Seq» впервые был упомянут в 2008 году. Количество рукописей, относящихся к RNA-Seq в названии или аннотация (рисунок, синяя линия) постоянно увеличивается с 6754 рукописями, опубликованными в 2018 году (ссылка на поиск PubMed ). Пересечение RNA-Seq и медицины (рисунок, золотая линия, ссылка на поиск PubMed ) имеет аналогичную скорость.
RNA-Seq имеет потенциал для выявления новой биологии болезни, профильных биомаркеров по клиническим показаниям, определения путей воздействия лекарств и постановки генетического диагноза. Эти результаты могут быть дополнительно персонализированы для подгрупп или даже отдельных пациентов, потенциально подчеркивая более эффективную профилактику, диагностику и терапию. Осуществимость этого подхода частично диктуется денежными и временными затратами; связанное с этим ограничение - необходимая группа специалистов (биоинформатики, врачи / клиницисты, базовые исследователи, техники) для полной интерпретации огромного количества данных, полученных в результате этого анализа.
Большое внимание было уделено данным RNA-Seq после того, как проекты Энциклопедии элементов ДНК (ENCODE) и Атлас генома рака (TCGA) использовали этот подход для характеристики десятков клеточных линий и тысячи образцов первичных опухолей соответственно. ENCODE направлен на идентификацию регуляторных областей всего генома в различных когортах клеточных линий, и транскриптомные данные имеют первостепенное значение для понимания последующего эффекта этих эпигенетических и генетических регуляторных слоев. TCGA, напротив, нацелен на сбор и анализ тысяч образцов пациентов из 30 различных типов опухолей, чтобы понять основные механизмы злокачественной трансформации и прогрессирования. В этом контексте данные RNA-Seq предоставляют уникальный снимок транскриптомного статуса заболевания и позволяют рассматривать беспристрастную популяцию транскриптов, что позволяет идентифицировать новые транскрипты, гибридные транскрипты и некодирующие РНК, которые можно было бы не обнаружить с помощью различных технологий.