
Войти
| Стилле реакция | |
|---|---|
| Названный в честь | Джон Кеннет Стилл |
| Тип реакции | Реакция сцепления |
| Идентификаторы | |
| Портал органической химии | ходовая часть |
| Идентификатор онтологии RSC | RXNO: 0000035 |
Реакция Стилле - это химическая реакция, широко используемая в органическом синтезе. Реакция включает связывание двух органических групп, одна из которых представляет собой оловоорганическое соединение (также известное как органостаннаны). Другим партнером по связыванию являются различные органические электрофилы. Реакция Стилле - одна из многих реакций сочетания, катализируемых палладием.
Группа R 1, присоединенная к триалкилолову, обычно sp 2 -гибридизована, включая винильные и арильные группы.
Эти органостаннаны также устойчивы как к воздуху, так и к влаге, и многие из этих реагентов либо коммерчески доступны, либо могут быть синтезированы на основании прецедентов в литературе. Однако эти реагенты с оловом очень токсичны. Х обычно представляет собой галогенид, например, Cl, Br или I, еще псевдогалогенидов, такие как трифлаты и сульфонаты и фосфаты также может быть использован. Опубликовано несколько обзоров.
Первый пример катализируемого палладием сочетания арилгалогенидов с оловоорганическими реагентами был описан Колином Иборном в 1976 году. Эта реакция давала от 7% до 53% диарильного продукта. Этот процесс был расширен до сочетания ацилхлоридов с реагентами алкил-олова в 1977 году компанией Toshihiko Migita, давая от 53% до 87% кетонового продукта.

В 1977 году опубликовала Migita дальнейшую работу над связью аллильных олова с реагентами как арил ( C) и ацильных ( D) галогенидов. Большая способность аллильных групп мигрировать на палладиевый катализатор позволяет проводить реакции при более низких температурах. Выходы арилгалогенидов составляют от 4% до 100%, а ацилгалогенидов - от 27% до 86%. Отражая ранний вклад Мигиты и Косуги, реакцию Стилле иногда называют сочетанием Мигиты – Косуги – Стилле.

Впоследствии Стилле сообщил в 1978 году о сочетании различных реагентов на основе алкилолова с многочисленными арил- и ацилгалогенидами в мягких условиях реакции с гораздо лучшими выходами (76% -99%). В 1980-х годах Стилле продолжил свою работу по синтезу множества кетонов с использованием этого широкого и мягкого процесса и выяснил механизм этого превращения.

К середине 1980-х было опубликовано более 65 статей на тему реакций сочетания с участием олова, в которых продолжалось изучение субстратного объема этой реакции. В то время как первоначальные исследования в этой области были сосредоточены на связывании алкильных групп, большая часть будущих работ включала гораздо более синтетически полезное соединение виниловых, алкенильных, арильных и аллилорганических станнанов с галогенидами. Благодаря устойчивости этих оловоорганических реагентов к воздуху и простоте их синтеза реакция Стилле стала обычным явлением в органическом синтезе.
Механизм реакции Стилле широко изучен. Каталитический цикл включает в себя окислительное добавление в виде галогенида или псевдогалоидом ( 2) к палладиевого катализатора ( 1), transmetalation из 3 с оловоорганического реагента ( 4), и восстановительного элиминирования из 5 с получением указанного в сочетании продукт ( 7), а также регенерированный палладиевый катализатор ( 1).

Однако детальный механизм связывания Стилле чрезвычайно сложен и может происходить через многочисленные реакционные пути. Как и другие реакции сочетания, катализируемые палладием, активный палладиевый катализатор, как полагают, представляет собой 14-электронный комплекс Pd (0), который можно получить различными способами. Использование 18- или 16-электронного источника Pd (0) Pd (PPh 3) 4, Pd (дБа) 2может подвергаться диссоциации лиганда с образованием активных частиц. Во- вторых, фосфины могут быть добавлены к ligandless палладий (0). Наконец, как показано на рисунке, восстановление источника Pd (II) ( 8) (Pd (OAc) 2, PdCl 2(MeCN) 2, PdCl 2(PPh 3) 2, BnPdCl (PPh 3) 2и т. д.) путем добавления фосфиновых лигандов или оловоорганических реагентов.
Предложено окислительное присоединение к 14-электронному комплексу Pd (0). Этот процесс дает 16-электронную разновидность Pd (II). Было высказано предположение, что анионные лиганды, такие как OAc, ускоряют этот этап за счет образования [Pd (OAc) (PR 3) n ] -, делая частицы палладия более нуклеофильными. В некоторых случаях, особенно при использовании sp 3 -гибридизованного галогенида, как правило, преобладает механизм типа S N 2, но это не так часто встречается в литературе. Однако, несмотря на то, что обычно образуется цис- промежуточное соединение после согласованного окислительного добавления, этот продукт быстро уравновешивается со своим транс- изомером.

Transmetalation из транса - промежуточного соединения из окислительного присоединения стадии, как полагает, протекает через различные механизмы в зависимости от субстратов и условий. Наиболее распространенный тип трансметалляции муфты Стилле включает ассоциативный механизм. Этот путь подразумевает, что органостаннан, обычно атом олова, связанный с аллильной, алкенильной или арильной группой, может координироваться с палладием через одну из этих двойных связей. Это дает мимолетные пятивалентные 18-электронные частицы, которые затем могут подвергаться отщеплению лиганда, снова образуя плоский квадратный комплекс. Несмотря на то, что органостаннан координируется с палладием через группу R 2, R 2 должен быть формально переведен на палладий (связь R 2 -Sn должна быть разорвана), а группа X должна уйти вместе с оловом, завершая трансметаллирование. Считается, что это происходит за счет двух механизмов.
Во-первых, когда станнан первоначально присоединяется к комплексу транс-металла, группа X может координироваться с оловом в дополнение к палладию, создавая циклическое переходное состояние. Разрушение этого аддукта приводит к потере R 3 Sn-X и трехвалентного палладиевого комплекса с R 1 и R 2, находящимися в цис- отношениях. Другой обычно наблюдаемый механизм включает такое же начальное добавление станнана к транс- палладиевому комплексу, как показано выше; однако в этом случае группа X не координируется с оловом, создавая открытое переходное состояние. После того, как α-углерод относительно олова атакует палладий, комплекс олова уйдет с чистым положительным зарядом. На схеме ниже обратите внимание, что двойная связь, координирующая олово, обозначает R 2, то есть любую алкенильную, аллильную или арильную группу. Кроме того, группа X может диссоциировать в любое время в течение механизма и связываться с комплексом Sn + в конце. Расчеты теории функциональной плотности предсказывают, что открытый механизм будет преобладать, если 2 лиганда останутся присоединенными к палладию, а группа X уйдет, тогда как циклический механизм более вероятен, если лиганд диссоциирует до трансметаллирования. Следовательно, хорошие уходящие группы, такие как трифлаты в полярных растворителях, благоприятствуют первым, тогда как объемные фосфиновые лиганды будут благоприятствовать вторым.

Менее распространенный путь трансметаллирования - диссоциативный механизм или механизм с участием растворителя. Здесь лиганд четырехвалентного палладия диссоциирует, и координирующий растворитель может добавляться к палладию. Когда растворитель отделяется с образованием трехвалентного промежуточного соединения с 14 электронами, станнан может присоединяться к палладию, подвергаясь процессу открытого или циклического типа, как указано выше.
Для того, чтобы R 1 -R 2 для восстановительно исключить эти группы должны занимать взаимно Cis координации сайтов. Следовательно, любые транс- аддукты должны изомеризоваться в цис- интермедиат, иначе связывание будет нарушено. Существует множество механизмов восстановительного устранения, и они обычно считаются согласованными.
Во-первых, четырехвалентный промежуточный продукт с 16 электронами, полученный на стадии трансметаллирования, может подвергаться самостоятельному восстановительному отщеплению из плоско-квадратного комплекса. Эта реакция протекает в два этапа: во-первых, за восстановительным элиминированием следует координация вновь образованной сигма-связи между R 1 и R 2 с металлом с окончательной диссоциацией с образованием связанного продукта.

Однако предыдущий процесс иногда является медленным и может быть значительно ускорен диссоциацией лиганда с образованием Т-образного интермедиата с 14 электронами. Этот промежуточный продукт может затем перегруппироваться с образованием Y-образного аддукта, который может подвергаться более быстрому восстановительному отщеплению.

Наконец, дополнительный лиганд может ассоциироваться с палладием с образованием 18-электронной тригонально-бипирамидной структуры с R 1 и R 2 цис друг к другу в экваториальных положениях. Геометрия этого промежуточного звена делает его похожим на Y-образную форму выше.

Наличие объемных лигандов также может увеличить скорость выведения. Лиганды, такие как фосфины, с большими углами прикуса вызывают стерическое отталкивание между L и R 1 и R 2, в результате чего угол между L и R группами увеличивается, а угол между R 1 и R 2, следовательно, уменьшается, что позволяет ускорить восстановительное удаление..

Скорость трансметаллирования органостаннанов с палладиевыми катализаторами показана ниже. Sp 2 -гибридизованные углеродные группы, присоединенные к олову, являются наиболее часто используемыми партнерами для связывания, а sp 3 -гибридизированные углеродные группы требуют более жестких условий, и концевые алкины могут быть связаны через связь CH посредством реакции Соногашира.

В качестве органического соединения олова обычно используют триметилстаннильное или трибутилстаннильное соединение. Хотя триметилстаннильные соединения демонстрируют более высокую реакционную способность по сравнению с трибутилстаннильными соединениями и имеют гораздо более простые спектры 1 H-ЯМР, токсичность первых намного выше.
Оптимизация того, какие лиганды лучше всего подходят для проведения реакции с высоким выходом и скоростью оборота, может быть трудной. Это связано с тем, что для окислительного добавления требуется металл, богатый электронами, следовательно, предпочтение отдается электронодонорным лигандам. Однако металл с дефицитом электронов более предпочтителен для стадий трансметаллирования и восстановительного удаления, что делает электроноакцепторные лиганды здесь лучшими. Следовательно, оптимальный набор лигандов сильно зависит от индивидуальных используемых субстратов и условий. Они могут изменить этап определения скорости, а также механизм этапа трансметаллирования.
Обычно используются лиганды с промежуточной донностью, такие как фосфины. Увеличение скорости можно увидеть при использовании лигандов с умеренным содержанием электронов, таких как три-2-фурилфосфин или трифениларсенин. Точно так же лиганды с большим числом доноров могут замедлять или ингибировать реакции связывания.
Эти наблюдения подразумевают, что обычно этапом, определяющим скорость реакции Стилле, является трансметаллирование.
Наиболее распространенной добавкой к реакции Стилле является стехиометрическая или сокаталитическая медь (I), в частности йодид меди, который может увеличить скорость более чем в 10 3 раз. Было высказано предположение, что в полярных растворителях медь трансметалатируется с помощью станнана. Полученный в результате органокупратный реагент может затем трансметаллировать с палладиевым катализатором. Кроме того, в эфирных растворителях медь также может способствовать удалению фосфинового лиганда, активируя центр Pd.
Было обнаружено, что хлорид лития является мощным ускорителем скорости в тех случаях, когда группа X диссоциирует от палладия (т. Е. Открытый механизм). Хлорид иона, как полагают, либо смещать X группу на палладий делает катализатор для более активного transmetalation или путем координации к Pd (0) аддукта для ускорения окислительного присоединения. Кроме того, LiCl, соль усиливает полярность растворителя, что делает его более легким для этого обычно анионного лиганда (- Cl, - Br, - OTf и т.д.), чтобы оставить. Эта добавка необходима, когда используется такой растворитель, как ТГФ ; однако использование более полярного растворителя, такого как NMP, может заменить потребность в этой солевой добавке. Однако, когда стадия трансметаллирования муфты протекает по циклическому механизму, добавление хлорида лития может фактически снизить скорость. Как и в циклическом механизме, нейтральный лиганд, такой как фосфин, должен диссоциировать вместо анионной группы X.
Наконец, на каталитический цикл также влияют источники фторид-ионов, такие как фторид цезия. Во-первых, фторид может увеличивать скорость реакции трифлаторганических соединений, возможно, за счет того же эффекта, что и хлорид лития. Кроме того, фторид - ионы могут выступать в качестве акцепторов для олова побочных продуктов, что делает их легче удалить с помощью фильтрации.
Наиболее распространенная побочная реакционная способность, связанная с реакцией Стилле, представляет собой гомосочетание реагентов станнана с образованием димера R 2 -R 2. Считается, что это происходит с помощью двух возможных механизмов. Во-первых, реакция двух эквивалентов станнана с предкатализатором Pd (II) после восстановительного элиминирования приведет к получению гомопаренного продукта. Во-вторых, катализатор Pd (0) может претерпеть радикальный процесс с образованием димера. Используемый органостаннановый реагент традиционно является четырехвалентным по олову и обычно состоит из переносимой sp 2 -гибридизированной группы и трех «непередаваемых» алкильных групп. Как видно выше, алкильные группы обычно медленнее всего мигрируют на палладиевый катализатор.

Также было обнаружено, что при температурах до 50 ° C арильные группы как на палладии, так и на координированном фосфине могут обмениваться. Хотя обычно они не обнаруживаются, во многих случаях они могут быть второстепенным продуктом.
Наконец, довольно редкая и экзотическая побочная реакция называется кинозамещением. Здесь, после начального окислительного присоединения в качестве арилгалогенида, этот вид Pd-Ar можно вставить через винил олово двойной связь. После отщепления β-гидрида, мигрирующего введения и протодестаннилирования может быть синтезирован 1,2-дизамещенный олефин.

Могут происходить многочисленные другие побочные реакции, в том числе E / Z-изомеризация, которая потенциально может быть проблемой при использовании алкенилстаннана. Механизм этой трансформации в настоящее время неизвестен. Обычно органостаннаны довольно устойчивы к гидролизу, но при использовании арилстаннанов, очень богатых электронами, это может стать значительной побочной реакцией.
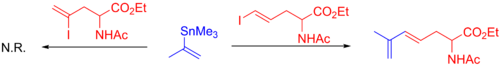
Винилгалогениды являются обычными партнерами по сочетанию в реакции Стилле, и реакции этого типа обнаруживаются в многочисленных полных синтезах природных продуктов. Обычно используются винилйодиды и бромиды. Винилхлориды недостаточно реактивны по отношению к окислительному присоединению к Pd (0). Обычно предпочтительны йодиды : они обычно реагируют быстрее и в более мягких условиях, чем бромиды. Это различие показано ниже на примере селективного связывания винилйодида в присутствии винилбромида.
Как правило, стереохимия из алкена сохраняется в течение всей реакции, за исключением в суровых условиях реакции. Могут использоваться различные алкены, в том числе α- и β-галоген-α, β-ненасыщенные кетоны, сложные эфиры и сульфоксиды (которые обычно нуждаются в добавке меди (I) для работы) и многое другое (см. Пример ниже).. Иногда используются также винилтрифлаты. Некоторые реакции требуют добавления LiCl, а другие замедляются, что означает наличие двух механистических путей.

Другой класс обычных электрофилов - арил- и гетероциклические галогениды. Что касается виниловых подложек, бромиды и йодиды встречаются чаще, несмотря на их большую стоимость. Можно выбрать множество арильных групп, включая кольца, замещенные электронодонорными заместителями, биарильные кольца и многое другое. Галогензамещенные гетероциклы также использовались в качестве партнеров для связывания, включая пиридины, фураны, тиофены, тиазолы, индолы, имидазолы, пурины, урацил, цитозины, пиримидины и многое другое (см. Ниже таблицу гетероциклов; галогены могут быть замещены в различных позиций по каждому).

Ниже приведен пример использования Stille связи со сложностью сборки на гетероциклов из нуклеозидов, таких как пуринов.

Арильные трифлаты и сульфонаты также пара для широкого спектра organostannane реагентов. Трифлаты имеют тенденцию реагировать сравнимо с бромидами в реакции Стилле.
Ацилхлориды также используются в качестве партнеров для связывания и могут использоваться с широким спектром органических станнанов, даже с реагентами алкил-олова, для получения кетонов (см. Пример ниже). Однако иногда сложно ввести ацилхлоридные функциональные группы в большие молекулы с чувствительными функциональными группами. Альтернативой, разработанной для этого процесса, является реакция перекрестного связывания с карбонилированием Стилле, при которой карбонильная группа вводится через введение моноксида углерода.

Также могут быть связаны аллильные, бензильные и пропаргиловые галогениды. Хотя обычно используются аллильные галогениды, они проходят через переходное состояние η 3, что позволяет связываться с органостаннаном в α- или γ-положении, преимущественно по наименее замещенному углероду (см. Пример ниже). Алкенильные эпоксиды (соседние эпоксиды и алкены ) также могут подвергаться такому же связыванию через переходное состояние η 3, что и открытие эпоксида для спирта. Хотя обычно используются аллиловые и бензиловые ацетаты, пропаргиловые ацетаты не реагируют с органостаннанами.

Органостаннановые реагенты являются общими. Некоторые из них имеются в продаже. Станнановые реагенты могут быть синтезированы реакцией реактива Гриньяра или литийорганического реагента с хлоридами триалкилолова. Например, винилтрибутилолово получают реакцией бромида винилмагния с хлоридом трибутилолова. Hydrostannylation из алкинов или алкенов предоставляет множество производных. Оловоорганические реагенты устойчивы к воздуху и влаге. Некоторые реакции могут происходить даже в воде. Их можно очистить хроматографией. Они толерантны к большинству функциональных групп. Некоторые оловоорганические соединения очень токсичны, особенно производные триметилстаннила.
Широко распространено использование реагентов на основе винилстаннана или алкенилстаннана. Что касается ограничений, как очень объемные реагенты на основе станнана, так и станнаны с замещением на α-углероде, как правило, реагируют вяло или требуют оптимизации. Например, в приведенном ниже случае α-замещенный винилстаннан реагирует только с концевым йодидом из-за стерических затруднений.
Арилстаннановые реагенты также широко распространены, и как электронодонорные, так и электроноакцепторные группы фактически увеличивают скорость трансметаллирования. Это снова означает, что могут иметь место два механизма трансметаллирования. Единственным ограничением для этих реагентов являются заместители в орто-положении, настолько малые, что метильные группы могут снизить скорость реакции. Широкое разнообразие гетероциклов (см. Раздел «Электрофилы») также можно использовать в качестве партнеров по связыванию (см. Пример с тиазольным кольцом ниже).


Алкинилстаннаны, наиболее реактивные из станнанов, также использовались в муфтах Стилле. Обычно они не нужны, поскольку концевые алкины могут напрямую связываться с палладиевыми катализаторами через свою связь CH через связывание Соногашира. Сообщалось, что аллилстаннаны работают, однако возникают трудности, как и с аллильными галогенидами, с трудностью контроля региоселективности для добавления α и γ. Реагенты дистаннан и ацилстаннан также использовались в муфтах Стилле.
Реакция Стилле использовалась в синтезе множества полимеров. Однако наиболее распространенным применением реакции Стилле является ее использование в органическом синтезе, в частности, в синтезе природных продуктов.
19-ступенчатый энантиоселективный полный синтез квадригема С Овермана включает реакцию двойного перекрестного метатезиса Стилле. Комплексный станнан присоединен к двум арилиодидным группам. Продукт получается после двойной циклизации Хека.

32 шаг Панек в энантиоселективного полный синтез из ansamycin антибиотиков (+) - mycotrienol использует поздней стадии тандемного типа Stille макроцикла муфты. Здесь органостаннан имеет две концевые группы трибутилолова, атакованные до алкена. Этот органостаннан «сшивает» два конца линейного исходного материала в макроцикл, добавляя в процесс два недостающих метиленовых звена. После окисления ароматического ядра с цериевым нитратом аммония (CAN) и удалением защитной группой с фтористоводородной кислотой дает натуральный продукт с выходом 54% в течение 3 -х этапов.

В 21 стадии энантиоселективного полного синтеза манзамина противоопухолевого алкалоида Ircinal A Стивена Ф. Мартина и его коллег используется тандемная реакция Стилла / Дильса-Альдера в одном сосуде. К винилбромиду добавляют алкеновую группу с последующим циклоприсоединением Дильса-Альдера in situ между добавленным алкеном и алкеном в пирролидиновом кольце.

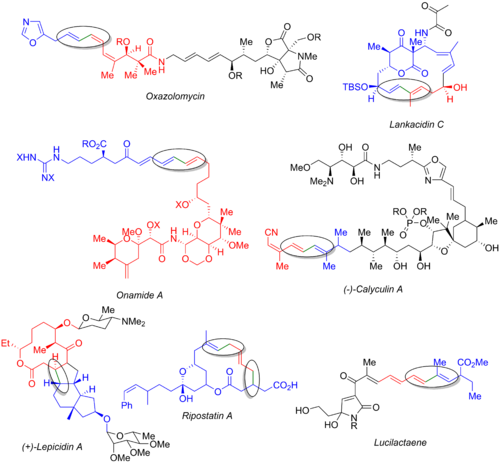
Многие другие общие синтезы используют реакцию Стилле, включая оксазоломицин, ланкацидин C, онамид A, каликулин A, лепицидин A, рипостатин A и люцилактаен. На изображении ниже показан конечный натуральный продукт : галогенид (синий), станнанорганический (красный) и образующаяся связь (зеленый и обведен). Из этих примеров ясно, что реакция Стилле может использоваться как на ранних стадиях синтеза (оксазоломицин и каликулин A), так и в конце конвергентного пути (онамид A, ланкацидин C, рипостатин A) или в средний (лепицидин А и люцилактаен). Синтез рипостатина A включает два одновременных связывания Стилле, за которыми следует метатезис с замыканием кольца. Синтез луцилактаена включает среднюю субъединицу, имеющую боран с одной стороны и станнан с другой, что позволяет провести реакцию Стилле с последующим сочетанием Сузуки.
Помимо проведения реакции в различных органических растворителях, были разработаны условия, которые позволяют использовать широкий спектр сочетаний Стилле в водном растворителе.
Было показано, что в присутствии солей Cu (I) палладий на угле является эффективным катализатором.
В области зеленой химии сообщается, что реакция Стилле происходит в низкоплавкой и высокополярной смеси сахара, такого как маннит, мочевина, такая как диметилмочевина, и соли, такой как хлорид аммония. Каталитическая система представляет собой трис (дибензилиденацетон) дипалладий (0) с трифениларсином :

Обычным изменением сочетания Стилле является включение карбонильной группы между R 1 и R 2, что служит эффективным методом образования кетонов. Этот процесс очень похож на первоначальное исследование Мигита и Стилле (см. «История») сочетания станнана с ацилхлоридами. Однако эти фрагменты не всегда легко доступны, и их может быть трудно образовать, особенно в присутствии чувствительных функциональных групп. Кроме того, может быть сложно контролировать их высокую реактивность. Взаимодействие Стилле-карбонилирование происходит в тех же условиях, что и сочетание Стилле, за исключением того, что используется атмосфера монооксида углерода (CO). CO может координироваться с палладиевым катализатором ( 9) после первоначального окислительного добавления с последующим введением CO в связь Pd-R 1 ( 10), что приводит к последующему восстановительному отщеплению до кетона ( 12). Этап трансметаллирования обычно является этапом определения скорости.

Ларри Оверман и его коллеги используют из Stille-carbonylative кросс-соединение в их 20-ступенчатом энантиоселективном полный синтез из стрихнина. Добавлял карбонил позже преобразуются в терминальный алкен с помощью реакции Виттига, что позволяет для ключа третичного азота и пятиядерного ядра, который будет сформирован через аза Сора - реакции Маннихи.
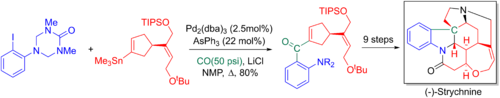
Джорджио Ортар и др. исследовал, как перекрестное связывание с карбонилированием Стилле может быть использовано для синтеза бензофеноновых фосфоров. Их встраивали в пептиды 4-бензоил-L-фенилаланина и использовали из-за их фотоаффинных свойств мечения для исследования различных взаимодействий пептид-белок.

16-ступенчатый полный рацемический синтез джатрафона Луи Хегедус включал перекрестное связывание Стилле-карбонилированием в качестве конечной стадии образования 11-членного макроцикла. Вместо галогенида в качестве связующего звена используется винилтрифлат.

Используя основополагающую публикацию Иборна в 1976 г., в которой арилстаннаны образуются из арилгалогенидов и дистаннанов, Т. Росс Келли применил этот процесс для внутримолекулярного связывания арилгалогенидов. Это тандемное сочетание станнилирование / арилгалогенид использовали для синтеза различных дигидрофенантренов. Большинство образующихся внутренних колец ограничено 5 или 6 членами, однако сообщалось о некоторых случаях макроциклизации. В отличие от обычного связывания Стилле, хлор не работает как галоген, возможно, из-за его более низкой реакционной способности в галогенной последовательности (его более короткая длина связи и более сильная энергия диссоциации связи затрудняют разрыв посредством окислительного присоединения ). Начиная с середины схемы ниже и двигаясь по часовой стрелке, палладиевый катализатор ( 1) окислительно присоединяется к наиболее реакционноспособной связи CX ( 13) с образованием 14 с последующим трансметаллированием с помощью дистаннана ( 15) с получением 16 и восстановительным элиминированием с получением арилстаннан ( 18). Регенерированный палладиевый катализатор ( 1) может окислительно добавляться ко второй связи CX 18 с образованием 19 с последующей внутримолекулярной трансметалляцией с получением 20 с последующим восстановительным элиминированием с получением связанного продукта ( 22).

Джи Джек Ли и др. использовали сочетание Стилла-Келли в своем синтезе различных кольцевых систем бензо [4,5] фуропиридинов. Они вызывают трехстадийный процесс, включающий аминирование Бухвальда-Хартвига, другую реакцию сочетания, катализируемую палладием, с последующим внутримолекулярным сочетанием Стилла-Келли. Обратите внимание, что арил-иодидная связь будет присоединяться к палладию в результате окисления быстрее, чем любая из арил-бромидных связей.
![Синтез бензо [4,5] фуропиридинов.](https://upload.wikimedia.org/wikipedia/commons/thumb/2/25/Benzofuropyridines.png/500px-Benzofuropyridines.png)
![{\ displaystyle {\ color {Blue} {\ ce {R ^ {1} -Sn (Alkyl) 3}}} + {\ color {Red} {\ ce {R ^ {2} -X}}} \ { \ ce {-gt; [{\ color {зеленый} {\ ce {Pd ^ {0}}}} {\ text {(каталитический)}}] [{\ text {набор лигандов}}]}} \ \ overbrace { {\ color {Blue} {\ ce {R ^ {1}}}} \! - \! {\ color {Red} {\ ce {R ^ {2}}}}} ^ {connected \ product} + { \ color {Red} {\ ce {X}}} \! - \! {\ color {Blue} {\ ce {Sn (алкил) 3}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/5baabb66db61c2d31fa2a5ca2b4e8156ee7c4133 "Общая схема реакции Стилле")
 : Аллил, алкенил, арил, бензил, ацил.
: Аллил, алкенил, арил, бензил, ацил. : галогениды (Cl, Br, I), псевдогалогениды ( OTf,), OAc
: галогениды (Cl, Br, I), псевдогалогениды ( OTf,), OAc