
Войти
 A реакция термит с использованием оксида железа (III). Вылетающие наружу искры представляют собой шарики расплавленного железа, сопровождаемые дымом.
A реакция термит с использованием оксида железа (III). Вылетающие наружу искры представляют собой шарики расплавленного железа, сопровождаемые дымом. A химическая реакция - это процесс, который приводит к химическому превращению одного набора химических веществ к другому. Классически химические реакции включают изменения, которые затрагивают только положения электронов в образовании и разрыве химических связей между атомами, без изменение на ядра (без изменений в присутствующих элементах), и часто может быть описано с помощью химического уравнения. Ядерная химия - это подраздел химии, который включает химические реакции нестабильных и радиоактивных элементов, где могут произойти как электронные, так и ядерные изменения.
Вещество (или вещества), первоначально участвующие в химической реакции, называются реагентами или реагентами. Химические реакции обычно характеризуются химическим изменением, и они дают один или несколько продуктов, которые обычно имеют свойства, отличные от реагентов. Реакции часто состоят из последовательности отдельных подэтапов, так называемых элементарных реакций, и информация о точном порядке действий является частью механизма реакции. Химические реакции описываются химическими уравнениями, которые символически представляют исходные материалы, конечные продукты, а иногда и промежуточные продукты и условия реакции.
Химические реакции происходят с характерной скоростью реакции при заданной температуре и химической концентрации. Обычно скорости реакции увеличиваются с повышением температуры, поскольку имеется больше тепловой энергии, доступной для достижения энергии активации, необходимой для разрыва связей между атомами.
Реакции могут продолжаться в прямом или обратном направлении, пока не дойдут до завершения или не достигнут равновесия. Реакции, которые идут в прямом направлении, чтобы приблизиться к равновесию, часто описываются как спонтанные, не требующие ввода свободной энергии для продвижения вперед. Несамопроизвольные реакции требуют ввода свободной энергии для продвижения вперед (примеры включают зарядку батареи с помощью внешнего источника электроэнергии или фотосинтез, вызванный поглощением электромагнитного излучения в форме солнечного света).
Различные химические реакции используются в комбинациях во время химического синтеза для получения желаемого продукта. В биохимии последовательные серии химических реакций (где продукт одной реакции является реагентом следующей реакции) образуют метаболические пути. Эти реакции часто катализируются белковыми ферментами. Ферменты увеличивают скорость биохимических реакций, так что метаболический синтез и разложение, невозможные в обычных условиях, могут происходить при температурах и концентрациях, присутствующих в клетке.
Общая концепция химической реакции была распространяется на реакции между объектами, меньшими, чем атомы, включая ядерные реакции, радиоактивные распады и реакции между элементарными частицами, как описано в квантовой теории поля.
 Антуан Лавуазье разработал теорию горения как химической реакции с кислородом.
Антуан Лавуазье разработал теорию горения как химической реакции с кислородом. Химические реакции, такие как горение в огонь, брожение и восстановление руд до металлов были известны с древних времен. Первоначальные теории преобразования материалов были разработаны греческими философами, например, Теория четырех элементов из Эмпедокла, утверждающая, что любое вещество состоит из четырех основных элементов - огня, воды, воздуха. и земля. В средневековье химические превращения изучали алхимики. Они пытались, в частности, преобразовать свинец в золото, для чего использовали реакции свинца и сплавов свинец-медь с серой.
Производство химических веществ которые обычно не встречаются в природе, уже давно опробованы, например, синтез серной и азотной кислоты, приписываемый неоднозначному алхимику Джабиру ибн Хайяну. Процесс включал нагревание сульфатных и нитратных минералов, таких как сульфат меди, квасцы и селитра. В 17 веке Иоганн Рудольф Глаубер произвел соляную кислоту и сульфат натрия путем взаимодействия серной кислоты и хлорида натрия. С развитием процесса со свинцовой камерой в 1746 году и процесса Леблана, позволяющего крупномасштабное производство серной кислоты и карбоната натрия, соответственно, химические реакции стали внедрен в промышленность. Дальнейшая оптимизация сернокислотной технологии привела к контактному процессу в 1880-х годах, а процесс Габера был разработан в 1909–1910 годах для синтеза аммиака.
С 16 века исследователи, в том числе Ян Баптист ван Гельмонт, Роберт Бойль и Исаак Ньютон, пытались создать теории экспериментально наблюдаемых химических превращений. Теория флогистона была предложена в 1667 году Иоганном Иоахимом Бехером. Он постулировал существование подобного огню элемента под названием «флогистон», который содержался в горючих телах и высвобождался во время сгорания. Это оказалось ложным в 1785 году Антуаном Лавуазье, который нашел правильное объяснение горения как реакции с кислородом из воздуха.
Жозеф Луи Гей-Люссак в 1808 году признал, что газы всегда реагируют. в определенных отношениях друг с другом. Основываясь на этой идее и атомарной теории Джона Далтона, Джозеф Пруст разработал закон определенных пропорций, который позже привел к концепции стехиометрия и химические уравнения.
Что касается органической химии, долгое время считалось, что соединения, полученные из живых организмов, слишком сложны для синтетического получения. Согласно концепции витализма, органическое вещество было наделено «жизненной силой» и отличалось от неорганических материалов. Однако это разделение было завершено синтезом мочевины из неорганических предшественников Фридрихом Велером в 1828 году. Другие химики, внесшие большой вклад в органическую химию, включают Александр Уильям Уильямсон с его синтезом простых эфиров и Кристофером Келком Ингольдом, который, среди многих открытий, установил механизмы реакций замещения.
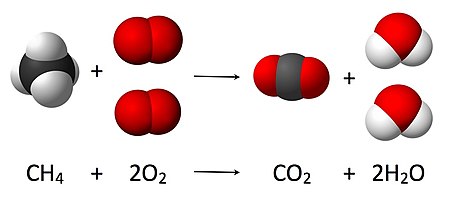 Как видно из уравнения CH. 4+ 2 O. 2→ CO. 2+ 2 H. 2O, перед газом кислородом на стороне реагентов необходимо поставить коэффициент 2 и перед водой на стороне продуктов, чтобы в соответствии с законом сохранения массы количество каждого элемента не изменялось во время реакции
Как видно из уравнения CH. 4+ 2 O. 2→ CO. 2+ 2 H. 2O, перед газом кислородом на стороне реагентов необходимо поставить коэффициент 2 и перед водой на стороне продуктов, чтобы в соответствии с законом сохранения массы количество каждого элемента не изменялось во время реакции Химические уравнения используются для графической иллюстрации химические реакции. Они состоят из химических или структурных формул реагентов слева и продуктов справа. Они разделены стрелкой (→), которая указывает направление и тип реакции; стрелка читается как слово «уступает». Острие стрелки указывает направление, в котором протекает реакция. Двойная стрелка (⇌), указывающая в противоположных направлениях, используется для равновесных реакций. Уравнения должны быть сбалансированы в соответствии со стехиометрией , количество атомов каждого вида должно быть одинаковым с обеих сторон уравнения. Это достигается путем масштабирования количества задействованных молекул (

Более сложные реакции представлены схемами реакций, которые, помимо исходных материалов и продуктов, показывают важные промежуточные соединения или переходные состояния. Кроме того, некоторые относительно незначительные добавки к реакции могут быть указаны над стрелкой реакции; примерами таких добавлений являются вода, тепло, освещение, катализатор и т. д. Аналогичным образом, некоторые второстепенные продукты могут быть помещены под стрелкой, часто с знак минус.
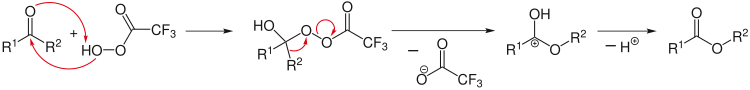 пример органической реакции: окисление кетонов до сложных эфиров с пероксикарбоновой кислотой
пример органической реакции: окисление кетонов до сложных эфиров с пероксикарбоновой кислотой ретросинтетический анализ может быть применен для создания комплекса реакция синтеза. Здесь анализ начинается с продуктов, например, путем расщепления выбранных химических связей, чтобы получить правдоподобные исходные реагенты. Специальная стрелка (⇒) используется в ретро-реакциях.
Элементарная реакция - это наименьшее звено, на которое может быть разложена химическая реакция, она имеет без промежуточных продуктов. Большинство экспериментально наблюдаемых реакций строятся из множества элементарных реакций, которые происходят параллельно или последовательно. Фактическая последовательность отдельных элементарных реакций известна как механизм реакции. В элементарной реакции участвует несколько молекул, обычно одна или две из-за малой вероятности того, что несколько молекул встретятся в определенное время.
 Изомеризация азобензола, вызванная светом (hν) или теплом ( Δ)
Изомеризация азобензола, вызванная светом (hν) или теплом ( Δ) Наиболее важными элементарными реакциями являются мономолекулярные и бимолекулярные реакции. Только одна молекула участвует в мономолекулярной реакции; он превращается путем изомеризации или диссоциации в одну или несколько других молекул. Такие реакции требуют добавления энергии в виде тепла или света. Типичным примером мономолекулярной реакции является цис-транс изомеризация, при которой цис-форма соединения превращается в транс-форму или наоборот.
В типичной реакции диссоциации связь в молекуле расщепляется (разрывается ), в результате чего образуются два молекулярных фрагмента. Расщепление может быть гомолитическим или гетеролитическим. В первом случае связь разделяется, так что каждый продукт сохраняет электрон и становится нейтральным радикалом. Во втором случае оба электрона химической связи остаются с одним из продуктов, в результате чего образуются заряженные ионы. Диссоциация играет важную роль в запуске цепных реакций, таких как реакции водород-кислород или полимеризация.

Для бимолекулярной>реакции две молекулы сталкиваются и вступают в реакцию друг с другом. Их слияние называется химический синтез или реакция присоединения.

Другая возможность состоит в том, что только часть одной молекулы передается другой молекуле. Этот тип реакции происходит, например, в окислительно-восстановительных и кислотно-основных реакциях. В окислительно-восстановительных реакциях переносимая частица представляет собой электрон, тогда как в кислотно-основных реакциях это протон. Этот тип реакции также называется метатезис.

например

реакции обратимы, то есть они могут протекать в обоих направлениях. Прямая и обратная реакции конкурируют друг с другом и отличаются скоростями реакции. Эти скорости зависят от концентрации и, следовательно, изменяются со временем реакции: обратная скорость постепенно увеличивается и становится равной скорости прямой реакции, устанавливая так называемое химическое равновесие. Время достижения равновесия зависит от таких параметров, как температура, давление и используемые материалы, и определяется минимальной свободной энергией. В состоянии равновесия свободная энергия Гиббса должна быть равна нулю. Зависимость от давления можно объяснить с помощью принципа Ле Шателье. Например, увеличение давления из-за уменьшения объема вызывает смещение реакции в сторону с меньшим количеством молей газа.
Выход реакции стабилизируется на уровне равновесия, но может быть увеличен за счет удаления продукта из реакции смеси или изменяют путем увеличения температуры или давления. Изменение концентраций реагентов не влияет на константу равновесия, но влияет на положение равновесия.
Химические реакции определяются законами термодинамики. Реакции могут происходить сами по себе, если они экзергонические, то есть если они выделяют энергию. Связанная свободная энергия реакции состоит из двух различных термодинамических величин: энтальпии и энтропии :
 .
.Реакции могут быть экзотермическими, где ΔH отрицательно и энергия выделяется. Типичными примерами экзотермических реакций являются осаждение и кристаллизация, в которых упорядоченные твердые вещества образуются из неупорядоченных газовых или жидких фаз. Напротив, в эндотермических реакциях тепло выделяется из окружающей среды. Это может происходить за счет увеличения энтропии системы, часто за счет образования газообразных продуктов реакции, которые имеют высокую энтропию. Поскольку энтропия увеличивается с температурой, многие эндотермические реакции предпочтительно протекают при высоких температурах. Напротив, многие экзотермические реакции, такие как кристаллизация, происходят при низких температурах. Изменения температуры иногда могут изменить знак энтальпии реакции, как в случае восстановления оксидом углерода диоксида молибдена :
 ;
; 
Эта реакция с образованием двуокиси углерода и молибден является эндотермическим при низких температурах, который становится меньше с повышением температуры. ΔH ° равен нулю при 1855 K, и реакция становится экзотермической выше этой температуры.
Изменения температуры также могут изменить направление реакции. Например, реакция конверсии водяного газа

предпочитается при низких температурах, но наоборот. высокой температурой. Смещение направления реакции происходит при 1100 К.
Реакции также можно охарактеризовать внутренней энергией, которая учитывает изменения энтропии, объема и химического потенциала. Последнее зависит, среди прочего, от активности задействованных веществ.

Скорость, с которой происходят реакции, изучается с помощью кинетики реакции. Скорость зависит от различных параметров, таких как:
. Некоторые теории позволяют рассчитать скорости реакции на молекулярном уровне. Это поле называется динамикой реакции. Скорость v реакции первого порядка, которая могла бы представлять собой распад вещества A, определяется как:
![{\ displaystyle v = - {\ гидроразрыв {d [{\ ce {A}}]} {dt}} = k \ cdot [{\ ce {A}}].}](https://wikimedia.org/api/rest_v1/media/math/render/svg/12291760fcaff20a02ff74abd0dfcb922664cddb)
Его интегрирование дает:
![{\ displaystyle {\ ce {[A]}} (t) = {\ ce {[A]}} _ {0} \ cdot e ^ {- k \ cdot t}.}](https://wikimedia.org/api/rest_v1/media/math/render/svg/498c37558508e2f7297604f93bb5408dcd8c3fd4)
Здесь k - первое- константа скорости порядка, имеющая размерность 1 / время, [A] (t) - концентрация в момент времени t, а [A] 0 - начальная концентрация. Скорость реакции первого порядка зависит только от концентрации и свойств задействованного вещества, а сама реакция может быть описана с помощью характеристического периода полураспада. При описании реакций более высокого порядка требуется более одной постоянной времени. Температурная зависимость константы скорости обычно соответствует уравнению Аррениуса :

где E a - это энергия активации, а k B - постоянная Больцмана. Одна из простейших моделей скорости реакции - теория столкновений. Более реалистичные модели адаптированы к конкретной проблеме и включают теорию переходного состояния, расчет поверхности потенциальной энергии, теорию Маркуса и Теория Райса – Рамспергера – Касселя – Маркуса (RRKM).
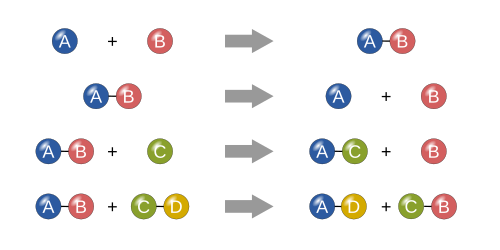 Представление четырех основных типов химических реакций: синтез, разложение, одиночная замена и двойная замена.
Представление четырех основных типов химических реакций: синтез, разложение, одиночная замена и двойная замена. В реакции синтеза два или более простых вещества объединяются, образуя более сложное вещество. Эти реакции имеют общую форму:

Два или более реагентов, дающих один продукт, - еще один способ идентифицировать синтез Одним из примеров реакции синтеза является комбинация железа и серы с образованием сульфида железа (II) :
Другой пример - простой газообразный водород в сочетании с простым газообразным кислородом для образования более сложного вещества, такого как вода.
Реакция разложения - это когда более сложное вещество распадается на более простые части. Таким образом, это противоположно реакции синтеза, и ее можно записать как

Одинпример разложения реакции - это электролиз воды с образованием кислорода и водорода в виде газа:
В реакции одиночной один несоединенный элемент заменяет другой в соединении; другими словами, Эти реакции имеют общую форму:

Один из реакций однократного замещения - это когда магний заменяет водород в воде, чтобы получить гидроксид магния и газообразный водород:

В реакции двойного замещения анионы и катионы меняются местами и образуют два совершенно разных соединения. Эти реакции имеют общую форму:

Например, когда барий хлорид>(BaCl 2) и сульфат магния (MgSO 4) реагируют, анион SO 4 переключается с 2Cl анион, давая соединение BaSO 4 и MgCl 2.
Другим примером реакции двойного воздействия нитрата свинца (II) с иодидом калия с образованием образуют иодид свинца (II) и нитрат калия :
В комбинации В соответствии с реакцией, элемент или соединение реагирует с кислородом, часто выделяется энергией в виде тепла или света. В реакциях горения всегда кислород, но также часто используется углеводород.

Реакция горения также может быть результатом углерода, магния. или сера, реагирующая с кислородом.


 Иллюстрация окислительно-восстановительной реакции
Иллюстрация окислительно-восстановительной реакции  Хлоридные соли образует в окислительно-восстановительной реакции металлического реакции окислительно-восстановительной реакции
Хлоридные соли образует в окислительно-восстановительной реакции металлического реакции окислительно-восстановительной реакции реакции окислительно-восстановительной реакции металлического >можно понять в условиях переноса электронов от одного взаимодействующего вещества (восстановитель ) к другому (окислитель ). В этом процессе первая разновидность окисляется, а вторая восстанавливается. Хотя этих описаний достаточно для многих целей, они не совсем верны. Окисление лучше определить как увеличение окисления, а восстановление как уменьшение степени уменьшения. Перенос электронов всегда приводит к окислительно-восстановительным процессам, которые вызывают «окислительно-восстановительные» (например, с участием ковалентных связей). В следующей окислительно-восстановительной реакции опасный металл натрия реагирует с токсичным газом хлором с образованием ионного соединения хлорида натрия или поваренной соли: В ходе реакции металлический натрий переходит в степень окисления от 0 (поскольку это чистый элемент) до +1: другими словами, натрий потерял один электрон и, как, окислился. элемент) к -1: хлор получает один электрон и его id был уменьшен. Когда хлор восстанавливается, он считается акцептором электронов или другими словами, окисление натрия - таким образом, газообразный хлор считается окислителем. Напротив, натрий окисляется или является донором электронов и таким образом, вызывает восстановление других частиц и считается восстановителем. Какой из задействованных реагентов будет восстановителем или окислителем, можно предсказать по электроотрицательности их элементов. Элементы с низкой электроотрицательностью, такими как большинство металлов, легко отдают электроны и окисляются - они восстанавливаются. Напротив, многие ионы с высокими степенями окисления, такие как H. 2O. 2, MnO. 4, CrO. 3, Cr. 2O. 7, OsO. 4, могут получить один или два дополнительных электрона и сильных окислителей. Число электронов, отданных или окислительно-восстановительной реакции, можно предсказать из конфигурации элемента-реагента. Элементы пытаются достичь низкоэнергетической конфигурации благородный газ, и поэтому щелочные металлы и галогены будут отдавать и принимать один электрон соответственно. Сами благородные газы химически неактивны. Важным классом окислительно-восстановительных сил используются электрохимические реакции, в качестве которых используются источники питания. Эти реакции особенно важны для производства химических элементов, таких как хлор или алюминий. Обратный процесс, в котором электроны высвобождаются окислительно-восстановительные реакторы.Комплексообразование
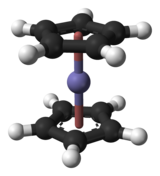 Ферроцен - атом железа, расположенный между двумя лигандами C 5H5
Ферроцен - атом железа, расположенный между двумя лигандами C 5H5
В реакциях комплексообразования несколько лигандов реагируют с атомом металла с образованием координационный комплекс. Это достигается предоставлением неподеленных пар лиганда на пустые орбитали атома металла и образования диполярных связей. Лиганды представляют собой основания Льюиса, они могут быть как ионами, так и нейтральными молекулами, такими как монооксид углерода, аммиак или вода. Количество лигандов, которые реагируют с центральным атомом металла, можно определить с помощью правил 18-электронов, согласно которому валентные оболочки переходного металла переходного металла будут коллективно вмещают 18 электронов, тогда как симметрию полученного комплекса можно предсказать с помощью теории кристаллического поля и теории поля лиганда. Реакции комплексообразования также включают обмен лиганда, в котором один или несколько лигандов заменяются другими, и окислительно-восстановительные процессы, которые изменяют степень окисления центрального атома металла.
В кислотно-основной теории Бренстеда - Лоури, кислотно-основная реакция включает перенос протонов (H) от одного вида (кислота ) к другому (основание ). Когда протон удаляется из кислоты, образующиеся частицы называют основанием, конъюгированным с этой кислотой,. Когда протон принимается основанием, образующиеся частицы называются конъюгированной кислотой этого основания. Другими словами, кислоты действуют как доноры протонов, а основания действуют как акцепторы протонов в соответствии со следующим уравнением:

Возможна обратная реакция, и, следовательно, кислота / основание и конъюгированное основание / кислоты всегда находятся в равновесии. Равновесие определяется константами диссоциации кислоты и основания (Kaи K b) вовлеченных веществ. Особым случаем кислотно-основной реакции является нейтрализация, где кислота и основание, взятые в одинаковых количествах, образуют нейтральную соль.
Кислотно-основные реакции могут иметь разные d определения в зависимости от используемой кислотно-щелочной концепции. Вот некоторые из наиболее распространенных:
 Осадки
Осадки Осадки - это образование твердого вещества в растворе или внутри другого твердого вещества во время химической реакции. Обычно это происходит, когда концентрация растворенных ионов превышает предел растворимости и образует нерастворимую соль. Этому процессу можно способствовать добавлением осаждающего агента или удалением растворителя. Быстрое осаждение приводит к аморфному или микрокристаллическому остатку, а медленный процесс может давать одиночные кристаллы. Последний также может быть получен путем перекристаллизации из микрокристаллических солей.
Реакции могут происходить между двумя твердыми веществами. Однако из-за относительно малых скоростей диффузии в твердых телах соответствующие химические реакции протекают очень медленно по сравнению с жидкостными и газофазными реакциями. Они ускоряются за счет повышения температуры реакции и тонкого разделения реагента для увеличения площади контактирующей поверхности.
Реакция может происходить на границе твердое тело | газ, поверхности при очень низком давлении, например сверхвысокий вакуум. С помощью сканирующей туннельной микроскопии можно наблюдать реакции на границе твердое тело | газ в реальном пространстве, если временная шкала реакции находится в правильном диапазоне. Реакции на границе твердое тело - газ в некоторых случаях связаны с катализом.
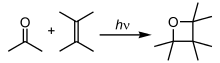 В этой реакции Патерно-Бюхи фотовозбужденная карбонильная группа добавляется к невозбужденному олефину, давая оксетан.
В этой реакции Патерно-Бюхи фотовозбужденная карбонильная группа добавляется к невозбужденному олефину, давая оксетан.В фотохимических реакциях атомы и молекулы поглощают энергию (фотоны ) освещающего света и переходят в возбужденное состояние. Затем они могут высвободить эту энергию, разрывая химические связи, тем самым производя радикалы. Фотохимические реакции включают реакции водород-кислород, радикальную полимеризацию, цепные реакции и реакции перегруппировки.
Многие важные процессы включают фотохимию. Главный пример - фотосинтез, в котором большинство растений используют солнечную энергию для преобразования углекислого газа и воды в глюкозу, удаляя кислород в качестве побочный продукт. Люди полагаются на фотохимию для образования витамина D, а зрение инициируется фотохимической реакцией родопсина. В светлячках фермент в брюшной полости катализирует реакцию, которая приводит к биолюминесценции. Многие важные фотохимические реакции, такие как образование озона, происходят в атмосфере Земли и составляют химический состав атмосферы.
 Схематическая диаграмма потенциальной энергии, показывающая действие катализатора в эндотермической химической реакции. Присутствие катализатора открывает другой путь реакции (красный) с более низкой энергией активации. Конечный результат и общая термодинамика совпадают.
Схематическая диаграмма потенциальной энергии, показывающая действие катализатора в эндотермической химической реакции. Присутствие катализатора открывает другой путь реакции (красный) с более низкой энергией активации. Конечный результат и общая термодинамика совпадают.  Твердые гетерогенные катализаторы наносят на сетки керамических каталитических нейтрализаторах, чтобы максимизировать их площадь поверхности. Этот выхлопных газов взят из Peugeot 106 S2 1100
Твердые гетерогенные катализаторы наносят на сетки керамических каталитических нейтрализаторах, чтобы максимизировать их площадь поверхности. Этот выхлопных газов взят из Peugeot 106 S2 1100 В реакция протекает не напрямую, а через реакцию с третьим веществом, известным как катализатор <397.>. Хотя катализатор принимает участие в реакции, к концу реакции он возвращается в исходное состояние и поэтому не расходуется. Однако он может быть заблокирован, деактивирован или уничтожен вторичными процессами. Катализаторы одна в другой фазе (гетерогенный ) или в той же фазе (гомогенный ), что и реагенты. В гетерогенном катализе типичные вторичные процессы включают коксование, когда катализатор покрывается полимерными побочными продуктами. Кроме того, гетерогенные катализаторы могут растворяться в системе твердое тело - жидкость или испаряться в системе твердое тело - газ. Катализаторы могут ускорить реакцию - химические вещества, замедляющие реакцию, называются ингибиторами. Вещества, повышающие активность катализаторов, называются промоторами, вещества, дезактивирующие катализаторы, называются каталитическими ядами. С помощью атомной реакции, которая кинетически ингибируется высокой энергией активации, может иметь место обход этой энергии активации.
Гетерогенные катализаторы обычно собой твердые частицы, измельченные в порошок для увеличения их площади. Особое значение в гетерогенном катализе металлы имеют платиновой группы и другие переходные металлы, которые используются в гидрогенизации, каталитическим риформинге и в синтезе промышленных химикатов, таких как азотная кислота и аммиак. Кислоты представляют собой пример гомогенного катализатора, они увеличивают нуклеофильность карбонилов, вызывающая реакция, которая иначе не протекала бы с электрофилами. Преимущество гомогенных катализаторов заключается в простоте их смешивания с реагентами, но их также может быть трудно отделить от продуктов. Поэтому во многих промышленных процессах предпочтение отдается гетерогенным катализаторам.
В органической химии, включая эффекты восстановления или кислотно-основных факторов, может протекать ряд других вариантов. место, которое включает ковалентные связи между атомами углерода или углерода и гетероатомами (такими как кислород, азот, галогены и т. д.). Многие специфические реакции в органической химии называют реакциями, названными в честь их первооткрывателей.
В реакции за ущерб, функциональная группа в конкретном химическом соединении заменяется другой группой. Эти реакции можно различать по типу замещающих веществ в нуклеофильном, электрофильном или механизме радикального замещения.
 SN1 механизм
SN1 механизм 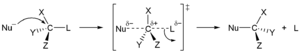 SN2 механизма
SN2 механизма В первом типа нуклеофил, атом или молекула с избытком электронов и, следовательно, с отрицательным зарядом или частичным зарядом, заменяет другой атом или часть молекулы «субстрата». Электронная пара от нуклеофила атакует субстрат, образуя новую связь, в то время как уходящая группа уходит с электронной парой. Нуклеофил может быть электрически нейтральным или заряженным отрицательно, тогда как субстрат обычно нейтрален или заряжен положительно. Примерами нуклеофилов являются ион гидроксида, алкоксиды, амины и галогениды. Этот тип часто встречается в основном в алифатических углеводородах и редко в ароматических углеводородах. Последние имеют высокую электронную плотность и включают в себя нуклеофильное ароматическое замещение только с очень сильными электроноакцепторными группами. Нуклеофильное замещение может происходить по двум различным механизмам: SN1 и SN2. В их названиях S означает замещение, N - нуклеофильное, а число представляет кинетический порядок реакции, мономолекулярный или бимолекулярный.
 Три стадии реакции SN2. Нуклеофил имеет зеленый цвет, а уходящая группа - красный.
Три стадии реакции SN2. Нуклеофил имеет зеленый цвет, а уходящая группа - красный.  SN2 реакция стереоинверсия (инверсия Вальдена)
SN2 реакция стереоинверсия (инверсия Вальдена) Реакция S N 1 протекает в два этапа. Во-первых, удаляемая группа удаляется, создаваемая карбокатион. За этим следует быстрая реакция с нуклеофилом.
В механизме S N 2 нуклеофил образует переходное состояние с атакованной молекулой, и только после этого уходящая группа отщепляется. Эти два механизма различаются стереохимией продуктов. S N 1 приводит к нестереоспецифическому добавлению и приводит не к хиральному центру, а скорее к набору геометрических изомеров (цис / транс). Напротив, в механизме S N 2 присутствует обращение (инверсия Вальдена ) ранее существовавшей стереохимии.
Электрофильное замещение является аналогом нуклеофильного замещения в том, что атакующий атом или молекула, электрофил, имеет низкую электронную плотность и, следовательно, положительный заряд. Типичными электрофилами являются атом углерода карбонильных групп, карбокатионы или катионы серы или нитрония. Эта реакция протекает исключительно в ароматических двигателях, где она электрофильным ароматическим заменителем. Электрофильная атака приводит к так называемому σ-комплексу, переходному состоянию, в котором ароматическая система исчезает. Затем отщепляющаяся группа, обычно протон, отщепляется и ароматичность восстанавливается. Альтернативой ароматическому замену является электрофильное алифатическое замещение. Он аналогичен нуклеофильному алифатическому замещению и имеет два основных типа: S E 1 и S E2
 Механизм электрофильного ароматического замещения
Механизм электрофильного ароматического замещения В реакции третьего типа за ущерб, радикального за ущерб, атакующие частица представляет собой радикал. Этот процесс обычно принимает форму цепной реакции, например, в реакции алканов с галогенами. На первом этапе свет или тепло разрушают галогенсодержащие молекулы, производящие радикалы. Затем реакция идет лавинообразно, пока два радикала не встретятся и не рекомбинируют.

добавление и его аналог, удаление, являются реакциями, которые определяют заместителей у атома углерода и образуют или разрывают кратные связи. Двойные и тройные связи могут быть получены путем удаления подходящей уходящей группы. Подобно нуклеофильному замещению, существует несколько возможных механизмов реакции, названных в честь соответствующего порядка реакции. В механизме E1 уходящая группа выбрасывается первой, образуя карбокатион. Следующая стадия, образование двойной связи, происходит с отщеплением протона (депротонирование ). В механизме E1cb порядок ухода меняется на противоположный, то есть сначала отщепляется протон. Этот механизм требует участия базы. Из-за схожих условий обе реакции в элиминировании E1 или E1cb всегда конкурируют с замещением S N 1.
 Элиминирование E1
Элиминирование E1  Удаление E1cb
Удаление E1cb  удаление E2
удаление E2 Механизм E2 также требует основания, но там атака основания и отщепление уходящей группы протекают одновременно и не образуют ионного промежуточного соединения. В отличие от отщеплений E1, для продукта реакции в механизме E2 возможны различные стереохимические конфигурации, поскольку атака основания предпочтительно происходит в анти-положении по отношению к уходящей группе. Из-за схожих условий и реагентов отщепление E2 всегда конкурирует с замещением S N 2.
Аналогом отщепления является добавление, где дважды или тройные связи преобразуются в одинарные. Подобно реакциям замещения, существует несколько типов добавок, различающихся по типу атакующей частицы. Например, при электрофильном присоединении бромистого водорода электрофил (протон) атакует двойную связь, образуя карбокатион, который затем вступает в реакцию с нуклеофилом (бромом). Карбокатион может быть образован по обе стороны от двойной связи в зависимости от групп, прикрепленных к его концам, и предпочтительную конфигурацию можно предсказать с помощью правила Марковникова. Это правило гласит, что «При гетеролитическом присоединении полярной молекулы к алкену или алкину более электроотрицательный (нуклеофильный) атом (или часть) полярной молекулы присоединяется к атому углерода, несущему меньшее количество атомов водорода».
Если присоединение функциональной группы происходит у менее замещенного атома углерода двойной связи, то электрофильное замещение кислотами невозможно. В этом случае необходимо использовать реакцию гидроборирования-окисления, где на первой стадии атом бора действует как электрофил и присоединяется к менее замещенному атому углерода. На втором этапе нуклеофильный гидропероксид или анион галогена атакует атом бора.
Хотя добавление к богатым электронами алкенам и алкинам в основном является электрофильным, нуклеофильное присоединение играет важную роль для кратных связей углерод-гетероатом, и особенно для его наиболее важного представителя, карбонильной группы. Этот процесс часто связан с отщеплением, так что после реакции карбонильная группа снова присутствует. Поэтому она называется реакцией присоединения-элиминирования и может происходить в производных карбоновых кислот, таких как хлориды, сложные эфиры или ангидриды. Эта реакция часто катализируется кислотами или основаниями, где кислоты увеличивают электрофильность карбонильной группы за счет связывания с атомом кислорода, тогда как основания усиливают нуклеофильность атакующего нуклеофила.
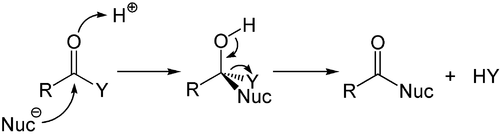 Катализируемый кислотой механизм присоединения-элиминирования
Катализируемый кислотой механизм присоединения-элиминирования Нуклеофильное присоединение карбаниона или другого нуклеофила к двойной связи альфа, бета ненасыщенного карбонильного соединения может происходить через Реакция Майкла, которая относится к большему классу добавок конъюгатов. Это один из наиболее эффективных методов мягкого образования связей C – C.
Некоторые добавки, которые не могут быть выполнены с нуклеофилами и электрофилами, могут быть заменены свободными радикалами. Как и в случае свободнорадикального замещения, радикальное присоединение протекает как цепная реакция, и такие реакции являются основой свободнорадикальной полимеризации.
 Механизм реакции Дильса-Альдера
Механизм реакции Дильса-Альдера  Перекрытие орбиталей в реакции Дильса-Альдера
Перекрытие орбиталей в реакции Дильса-Альдера В реакции перегруппировки углеродный скелет молекула перегруппирована, чтобы дать структурный изомер исходной молекулы. К ним относятся реакции гидридного сдвига, такие как перегруппировка Вагнера-Меервейна, где водород, алкил или арил группа мигрирует от одного углерода к соседнему. Большинство перегруппировок связано с разрывом и образованием новых углерод-углеродных связей. Другими примерами являются сигматропная реакция, такая как перегруппировка Коупа.
Циклические перегруппировки включают циклоприсоединения и, в более общем смысле, перициклические реакции, где две или более молекулы, содержащие двойную связь, образуют циклическую молекулу. Важным примером реакции циклоприсоединения является реакция Дильса – Альдера (так называемое [4 + 2] циклоприсоединение) между сопряженным диеном и замещенным алкеном для образования замещенной системы циклогексена.
Будет ли происходить определенное циклоприсоединение, зависит от электронных орбиталей участвующих частиц, поскольку только орбитали с тем же знаком волновой функции будут пересекаться и конструктивно взаимодействовать для формирования новых связей. Циклоприсоединение обычно осуществляется с помощью света или тепла. Эти возмущения приводят к различному расположению электронов в возбужденном состоянии вовлеченных молекул и, следовательно, к различным эффектам. Например, реакциям [4 + 2] Дильса-Альдера может способствовать тепло, тогда как [2 + 2] циклоприсоединение селективно индуцируется светом. Из-за орбитального характера потенциал для образования стереоизомерных продуктов при циклоприсоединении ограничен, как описано правилами Вудворда-Хоффмана.
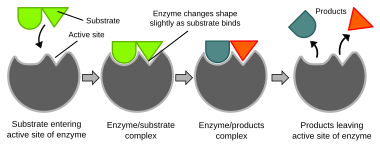 Иллюстрация модели индуцированного соответствия фермента активность
Иллюстрация модели индуцированного соответствия фермента активность Биохимические реакции в основном контролируются ферментами. Эти белки могут специфически катализировать единственную реакцию, так что можно очень точно контролировать реакции. Реакция происходит в активном сайте, небольшой части фермента, которая обычно находится в щели или кармане, выстланном аминокислотными остатками, а остальная часть фермента используется в основном для стабилизации. Каталитическое действие ферментов зависит от нескольких механизмов, включая форму молекулы («индуцированное соответствие»), деформацию связи, близость и ориентацию молекул относительно фермента, донорство или удаление протонов (кислотный / основной катализ), электростатические взаимодействия и многие другие.
Биохимические реакции, происходящие в живых организмах, в совокупности известны как метаболизм. Среди наиболее важных его механизмов - анаболизм, при котором различные ДНК и процессы, контролируемые ферментами, приводят к производству больших молекул, таких как белки и углеводы из более мелких единиц. Биоэнергетика изучает источники энергии для таких реакций. Важным источником энергии является глюкоза, которая может вырабатываться растениями посредством фотосинтеза или ассимилироваться с пищей. Все организмы используют эту энергию для производства аденозинтрифосфата (АТФ), который затем можно использовать для активации других реакций.
 Реакция термитов при сварке железных дорог. Вскоре после этого жидкое железо течет в кристаллизатор вокруг зазора рельса.
Реакция термитов при сварке железных дорог. Вскоре после этого жидкое железо течет в кристаллизатор вокруг зазора рельса. Химические реакции являются центральными в химической инженерии, где они используются для синтеза новых соединений из природного сырья, такого как нефтяные и минеральные руды. Важно сделать реакцию максимально эффективной, максимизировать выход и минимизировать количество реагентов, энергозатрат и отходов. Катализаторы особенно полезны для уменьшения энергии, необходимой для реакции, и увеличения ее скорости реакции.
У некоторых конкретных реакций есть свои нишевые применения. Например, реакция термит используется для генерации света и тепла в пиротехнике и сварке. Хотя она менее управляема, чем более традиционная кислородно-топливная сварка, дуговая сварка и сварка оплавлением, она требует гораздо меньше оборудования и по-прежнему используется для ремонта рельсов., особенно в отдаленных районах.
Механизмы мониторинга химических реакций сильно зависят от скорости реакции. Относительно медленные процессы могут быть проанализированы на месте на предмет концентраций и идентичности отдельных ингредиентов. Важными инструментами анализа в реальном времени являются измерение pH и анализ спектров оптического поглощения (цвета) и излучения. Менее доступный, но довольно эффективный метод - введение радиоактивного изотопа в реакцию и наблюдение за тем, как он изменяется с течением времени и куда он движется; этот метод часто используется для анализа перераспределения веществ в организме человека. Более быстрые реакции обычно изучаются с помощью сверхбыстрой лазерной спектроскопии, где использование фемтосекунд лазеров позволяет отслеживать короткоживущие переходные состояния во времени, уменьшенном до нескольких фемтосекунд.
| Викицитатник содержит цитаты, относящиеся к: Химическая реакция |
