
Войти
Литиевоорганические реагенты - это металлоорганические соединения, содержащие углерод - литиевые связи. Они являются важными реагентами в органическом синтезе и часто используются для переноса органической группы или атома лития на субстраты на стадиях синтеза посредством нуклеофильного присоединения или простого депротонирования. Литийорганические реагенты используются в промышленности в качестве инициатора анионной полимеризации, которая приводит к получению различных эластомеров. Они также применялись в асимметричном синтезе в фармацевтической промышленности. Из-за большой разницы в электроотрицательности между атомом углерода и атомом лития связь C-Li является сильно ионной. Из-за полярной природы связи C-Li литийорганические реагенты являются хорошими нуклеофилами и сильными основаниями. Для лабораторного органического синтеза многие литийорганические реагенты коммерчески доступны в виде растворов. Эти реагенты обладают высокой реакционной способностью и иногда пирофорными.
 Стеклянными бутылями, содержащими бутиллитий
Стеклянными бутылями, содержащими бутиллитий  втор-бутиллитиевый агрегат
втор-бутиллитиевый агрегат Исследования литийорганических реагентов начались в 1930-х годах и были впервые проведены K Арл Циглер, Георг Виттиг и Генри Гилман. По сравнению с реагентами Гриньяра (магний), литийорганические реагенты часто могут выполнять одни и те же реакции с повышенными скоростями и более высокими выходами, например, в случае металлирования. С тех пор литийорганические реагенты в общем использовании обогнали реагенты Гриньяра.
Хотя простые разновидности алкиллития часто представлены как мономер RLi, они существуют в виде агрегатов (олигомеры ) или полимеры. Степень агрегации зависит от органического заместителя и присутствия других лигандов. Эти структуры были выяснены различными методами, в частности с помощью спектроскопии ЯМР Li, Li и C и рентгеноструктурного анализа. Вычислительная химия подтверждает эти предположения.
 Делокализованная электронная плотность в аллиллитиевых реагентах
Делокализованная электронная плотность в аллиллитиевых реагентах Относительные электроотрицательности углерода и лития предполагают, что связь C-Li будет сильно полярной. Однако некоторые литийорганические соединения обладают такими свойствами, как растворимость в неполярных растворителях, что усложняет проблему. В то время как большинство данных предполагают, что связь C-Li является по существу ионной, ведутся споры о том, существует ли небольшой ковалентный характер в связи C-Li. Согласно одной оценке процент ионного характера соединений алкиллития составляет от 80 до 88%.
В соединениях аллиллития катион лития координируется с лицевой стороной углеродной π-связи по η-моде вместо локализованного карбанионного центр, таким образом, аллиллитий часто менее агрегирован, чем алкиллитий. В комплексах ариллития катион лития координируется с одним карбанионным центром через связь Li-C σ-типа.


 Твердотельные структуры тетрамеров метиллития, гексамеров н-бутиллития и полимерной лестницы фениллития
Твердотельные структуры тетрамеров метиллития, гексамеров н-бутиллития и полимерной лестницы фениллития  Металлические ядра тетраэдра и октаэдра, образованные агрегацией координационного комплекса треугольник Li3 - карбанион
Металлические ядра тетраэдра и октаэдра, образованные агрегацией координационного комплекса треугольник Li3 - карбанион Подобно другим видам, состоящим из полярных субъединиц, литийорганические соединения объединяются. На формирование агрегатов влияют электростатические взаимодействия, координация между литием и окружающими молекулами растворителя или полярными добавками, а также стерические эффекты.
Основным строительным блоком для создания более сложных структур является карбанионный центр. взаимодействуют с треугольником Li 3 по типу η- 3. В простых алкиллитиевых реагентах эти треугольники объединяются с образованием тетраэдрических или октаэдрических структур. Например, метиллитий и трет-бутиллитий все присутствуют в тетрамере [RLi] 4. Метиллитий существует в виде тетрамеров в кластере кубанового типа в твердом состоянии, с четырьмя центрами лития, образующими тетраэдр. Каждый метанид в тетрамере в метиллитии может иметь агостическое взаимодействие с катионами лития в соседних тетрамерах. С другой стороны, этиллитий и трет-бутиллитий не проявляют этого взаимодействия и поэтому растворимы в неполярных углеводородных растворителях. Другой класс алкиллития имеет гексамерные структуры, такие как н-бутиллитий, изопропиллитий и циклогексаниллитий.
 Димер LDA с ТГФ, координированный с катионами Li
Димер LDA с ТГФ, координированный с катионами Li Обычные амиды лития, например бис (триметилсилил) амид лития и диизопропиламид лития также подвержены агрегации. Амиды лития принимают структуры типа полимерной лестницы в некоординирующем растворителе в твердом состоянии и обычно существуют в виде димеров в эфирных растворителях. В присутствии сильно донорных лигандов образуются три- или тетрамерные литиевые центры. Например, LDA существует в основном в виде димеров в THF. Структуры обычных амидов лития, таких как диизопропиламид лития (LDA) и гексаметилдисилазид лития (LiHMDS), были тщательно изучены Коллумом и его сотрудниками с использованием ЯМР-спектроскопии. Другой важный класс реагентов - это силиллитий, который широко используется в синтезе металлоорганических комплексов и полисилановых дендримеров. В твердом состоянии, в отличие от алкиллитиевых реагентов, большинство силиллитий имеет тенденцию к образованию мономерных структур, координированных с молекулами растворителя, такими как ТГФ, и только несколько силиллитий были охарактеризованы как высшие агрегаты. Это различие может возникать из-за способа получения силиллитий, стерических затруднений, вызванных объемными алкильными заместителями на кремнии, и менее поляризованной природы связей Si-Li. Добавление сильно донорных лигандов, таких как TMEDA и (-) - спартеин, может вытеснять молекулы координирующего растворителя в силиллитиях.
Опираясь исключительно на структурную Информация об агрегатах литийорганического происхождения, полученных в твердом состоянии из кристаллических структур, имеет определенные пределы, поскольку литийорганические реагенты могут принимать различные структуры в среде реакционного раствора. Кроме того, в некоторых случаях трудно выделить кристаллическую структуру литийорганического соединения. Следовательно, изучение структуры литийорганических реагентов и литийсодержащих промежуточных продуктов в форме раствора чрезвычайно полезно для понимания реакционной способности этих реагентов. ЯМР-спектроскопия стала мощным инструментом для исследования агрегатов лития в растворе. Для алкиллитиевых соединений соединение C-Li J часто можно использовать для определения количества лития, взаимодействующего с карбанионным центром, а также для определения того, являются ли эти взаимодействия статическими или динамическими. Отдельные сигналы ЯМР могут также отличать присутствие нескольких агрегатов от общей мономерной единицы.
На структуру литийорганических соединений влияет присутствие оснований Льюиса, таких как тетрагидрофуран (THF), диэтиловый эфир (Et 2 O), тетраметилэтилендиамин (TMEDA) или гексаметилфосфорамид (HMPA). Метиллитий представляет собой особый случай, в котором сольватация эфиром или полярной добавкой HMPA не дезагрегирует тетрамерную структуру в твердом состоянии. С другой стороны, ТГФ дезагрегирует гексамерный бутиллитий: тетрамер является основным компонентом, а ΔG для взаимного превращения между тетрамером и димером составляет около 11 ккал / моль. TMEDA может также образовывать хелатные комплексы с катионами лития в н-бутиллитии и образовывать сольватированные димеры, такие как [(TMEDA) LiBu-n)] 2. Было показано, что фениллитий существует в виде искаженного тетрамера в кристаллизованном эфирном сольвате и в виде смеси димера и тетрамера в эфирном растворе.
| Растворитель | Структура | |
|---|---|---|
| метиллитий | THF | тетрамер |
| метиллитий | эфир / HMPA | тетрамер |
| н-бутиллитий | пентан | гексамер |
| н-бутиллитий | эфир | тетрамер |
| н-бутиллитий | THF | тетрамер-димер |
| втор-бутиллитий | пентан | гексамер-тетрамер |
| изопропиллитий | пентан | гексамер-тетрамер |
| трет-бутиллитий | пентан | тетрамер |
| трет-бутиллитий | ТГФ | мономер |
| фениллитий | эфир | тетрамер-димер |
| фениллитий | эфир / HMPA | димер |
Поскольку структура литийорганических реагентов изменяется в зависимости от их химического окружения, меняется и их реактивность. ты и избирательность. Один из вопросов, касающихся взаимосвязи структура-реакционная способность, заключается в том, существует ли корреляция между степенью агрегации и реакционной способностью литийорганических реагентов. Первоначально предполагалось, что низшие агрегаты, такие как мономеры, более реакционноспособны в алкиллитии. Однако пути реакций, в которых реакционноспособными частицами являются димеры или другие олигомеры, также были обнаружены, а для амидов лития, таких как LDA, реакции на основе димеров являются обычными. Серия исследований кинетики раствора LDA-опосредованных реакций предполагает, что более низкие агрегаты енолятов не обязательно приводят к более высокой реакционной способности.
Кроме того, некоторые основания Льюиса повышают реакционную способность литийорганических соединений. Однако не всегда ясно, действуют ли эти добавки как сильные хелатирующие лиганды и как наблюдаемое повышение реакционной способности связано со структурными изменениями в агрегатах, вызванными этими добавками. Например, TMEDA увеличивает скорость и эффективность многих реакций с участием литийорганических реагентов. Что касается алкиллитиевых реагентов, TMEDA действует как лиганд-донор, снижает степень агрегации и увеличивает нуклеофильность этих веществ. Однако TMEDA не всегда действует как лиганд-донор для катиона лития, особенно в присутствии анионных кислородных и азотных центров. Например, он слабо взаимодействует с LDA и LiHMDS даже в углеводородных растворителях без конкурирующих донорных лигандов. При литировании имина, в то время как ТГФ действует как сильный донорный лиганд для LiHMDS, слабо координирующий TMEDA легко диссоциирует от LiHMDS, что приводит к образованию димеров LiHMDS, которые являются более реактивными частицами. Таким образом, в случае LiHMDS TMEDA не увеличивает реакционную способность за счет снижения агрегационного состояния. Кроме того, в отличие от простых соединений алкиллития, TMEDA не дезагрегирует литиоацетофенолят в растворе ТГФ. Добавление HMPA к амидам лития, таким как LiHMDS и LDA, часто приводит к смеси агрегатов димер / мономер в ТГФ. Однако соотношение разновидностей димера / мономера не изменяется с увеличением концентрации HMPA, таким образом, наблюдаемое повышение реакционной способности не является результатом дезагрегации. Механизм увеличения реакционной способности этих добавок все еще исследуется.
Связь C-Li в литийорганических реагентах сильно поляризована. В результате углерод притягивает большую часть электронной плотности в связи и напоминает карбанион. Таким образом, литийорганические реагенты являются сильно основными и нуклеофильными. Некоторые из наиболее распространенных применений литийорганических реагентов в синтезе включают их использование в качестве нуклеофилов, сильных оснований для депротонирования, инициатора полимеризации и исходного материала для получения других металлоорганических соединений.
В качестве нуклеофилов литийорганические реагенты претерпевают реакции карболитирования, в результате чего связь углерод-литий присоединяется через двойную или тройную связь углерод-углерод, образуя новые литийорганические соединения. Эта реакция является наиболее широко применяемой реакцией литийорганических соединений. Карболитирование является ключевым в процессах анионной полимеризации, и н-бутиллитий используется в качестве катализатора для инициирования полимеризации стирола, бутадиена или изопрена или их смесей.

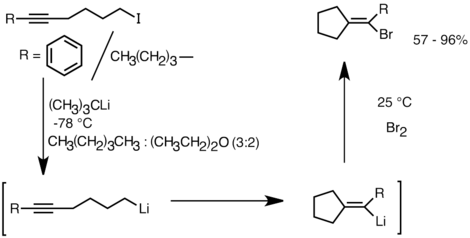
Другое применение, которое Преимущество этой реакционной способности заключается в образовании карбоциклических и гетероциклических соединений посредством внутримолекулярного карболитирования. Как форма анионной циклизации, реакции внутримолекулярного карболитирования имеют несколько преимуществ перед радикальной циклизацией. Во-первых, образующиеся циклические литийорганические соединения могут реагировать с электрофилами, в то время как часто бывает трудно уловить радикальный промежуточный продукт соответствующей структуры. Во-вторых, анионные циклизации часто более регио- и стереоспецифичны, чем радикальная циклизация, особенно в случае 5-гексениллитий. Внутримолекулярное карболитирование позволяет присоединять алкил-, виниллитий к тройным связям и моноалкилзамещенным двойным связям. Ариллитий также может присоединяться, если образуется 5-членное кольцо. Ограничения внутримолекулярного карболитирования включают сложность образования 3- или 4-членных колец, поскольку промежуточные циклические органические соединения лития часто имеют тенденцию к раскрытию кольца. Ниже приведен пример реакции внутримолекулярного карболитирования. Виды лития, полученные в результате литий-галогенового обмена, циклизовались с образованием виниллития за счет замыкания 5-экзо-триггерного кольца. Соединения виниллития далее реагируют с электрофилами и производят функционализированные циклопентилиденовые соединения.
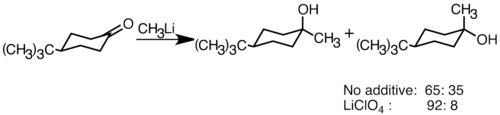
Нуклеофильные литийорганические реагенты могут добавляться к электрофильным двойным карбонильным связям с образованием углерод-углеродных связей. Они могут реагировать с альдегидами и кетонами с образованием спиртов. Добавление происходит в основном через полярное добавление, при котором нуклеофильные органические соединения лития атакуют с экваториального направления и производят аксиальный спирт. Добавление солей лития, таких как LiClO 4, может улучшить стереоселективность реакции.
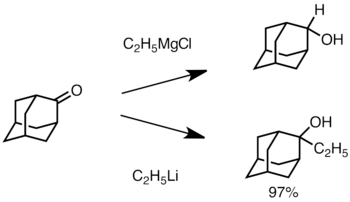
Когда кетон является стерически затрудненным, использование реактивов Гриньяра часто приводит к восстановлению карбонильной группы вместо добавления. Однако алкиллитиевые реагенты с меньшей вероятностью восстанавливают кетон и могут быть использованы для синтеза замещенных спиртов. Ниже приведен пример добавления этиллития к адамантону для получения третичного спирта.
Литиевоорганические реагенты также лучше, чем реагенты Гриньяра, по своей способности реагировать с карбоновыми кислотами с образованием кетонов. Эту реакцию можно оптимизировать, тщательно контролируя количество добавляемого литийорганического реагента или используя триметилсилилхлорид для гашения избытка литиевого реагента. Более распространенный способ синтеза кетонов - добавление литийорганических реагентов к амидам Вайнреба (N-метокси-N-метиламиды). Эта реакция дает кетоны, когда литийорганические реагенты используются в избытке из-за хелатирования иона лития между N-метокси кислородом и карбонильным кислородом, который образует тетраэдрический промежуточный продукт, который разрушается при кислотной обработке.

Литиевые реагенты также могут реагируют с диоксидом углерода с образованием карбоновых кислот.
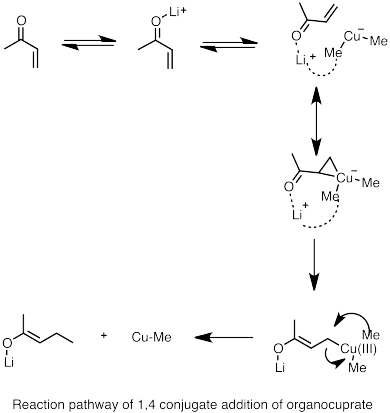
В случае еноновых субстратов, где возможны два места нуклеофильного присоединения (1,2 присоединение к карбонильному углероду или 1,4 присоединение конъюгата к β-углероду), наиболее реакционноспособные литийорганические соединения предпочитают 1,2-присоединение, однако существует несколько способов заставить литийорганические реагенты подвергаться добавлению конъюгата. Во-первых, поскольку 1,4-аддукт, вероятно, будет более термодинамически благоприятным видом, добавление конъюгата может быть достигнуто путем уравновешивания (изомеризация двух продуктов), особенно когда нуклеофил лития является слабым и 1,2-присоединение является обратимым. Во-вторых, добавление донорных лигандов к реакции образует стабилизированные гетероатомами литиевые частицы, которые способствуют присоединению 1,4-конъюгата. В одном примере добавление ГМПА в низком уровне к растворителю способствует добавлению 1,4. В отсутствие донорного лиганда катион лития тесно координирован с атомом кислорода, однако, когда катион лития сольватируется HMPA, координация между кислородом карбонила и ионом лития ослабляется. Этот метод обычно не может использоваться для воздействия на региоселективность алкил- и ариллитиевых реагентов.

Литиевые реагенты могут также выполнять энантиоселективное нуклеофильное присоединение к карбонилу и его производным, часто в присутствии хиральных лигандов. Эта реакционная способность широко применяется в промышленном синтезе фармацевтических соединений. Примером является синтез «Мерк энд Дюпон» эфавиренца, мощного ингибитора обратной транскриптазы ВИЧ. Ацетилид лития добавляют к прохиральному кетону с получением хирального спирта. Структура активного промежуточного продукта реакции была определена с помощью исследований ЯМР-спектроскопии в состоянии раствора и рентгеновской кристаллографии твердого состояния как кубический тетрамер 2: 2.

Литийорганические реагенты могут служат в качестве нуклеофилов и проводят реакции типа S N 2 с алкильными или аллильными галогенидами. Хотя они считаются более реактивными, чем реакции Гриньяра при алкилировании, их использование все еще ограничено из-за конкурирующих побочных реакций, таких как радикальные реакции или обмен металл-галоген. Большинство литийорганических реагентов, используемых при алкилировании, являются более стабилизированными, менее основными и менее агрегированными, например, реагенты, стабилизированные гетероатомом, арил- или аллиллитиевые реагенты. Было показано, что HMPA увеличивает скорость реакции и выход продукта, а реакционная способность ариллитиевых реагентов часто повышается за счет добавления алкоксидов калия. Литиевые реагенты также могут выполнять нуклеофильные атаки с эпоксидами с образованием спиртов.

литийорганические реагенты обеспечивают широкий диапазон основности. трет-бутиллитий с тремя слабо электронодонорными алкильными группами, является самым сильным основанием, имеющимся в продаже (pKa = 53). В результате кислотные протоны на -ОН, -NH и -SH часто защищены в присутствии литийорганических реагентов. Некоторые обычно используемые литиевые основания представляют собой разновидности алкиллития, такие как н-бутиллитий и диалкиламиды лития (LiNR 2). Реагенты с объемными R-группами, такие как диизопропиламид лития (LDA) и бис (триметилсилил) амид лития (LiHMDS), часто стерически затруднены для нуклеофильного присоединения и, таким образом, более избирательны в отношении депротонирования. Диалкиламиды лития (LiNR 2) широко используются в образовании енолята и альдольной реакции. На реакционную способность и селективность этих оснований также влияют растворители и другие противоионы.
Металлизация с помощью литийорганических реагентов, также известная как литиирование или литий-водородный обмен, достигается, когда литийорганический реагент, чаще всего алкиллитий, извлекает протон и образует новый органолитий.
 | (1) |
Обычными реагентами для металлирования являются бутиллитий. Трет-Бутиллитий и втор-бутиллитий, как правило, более реакционноспособны и имеют лучшую селективность, чем н-бутиллитий, однако они также более дороги и сложны в обращении. Металлирование - это распространенный способ получения универсального литийорганического соединения. реагентов. Положение металлирования в основном контролируется кислотностью связи CH. Литирование часто происходит в положении α к электроноакцепторным группам, так как они хорошо стабилизируют электронную плотность аниона. группы на ароматических соединениях и гетероциклах обеспечивают региоселективные участки металлирования; направленное орто-металлирование является важным классом реакций металлирования. Металлизированные сульфоны, ацил g Группы и α-металлированные амиды являются важными промежуточными продуктами в химическом синтезе. Металлизация аллилового эфира алкиллитием или LDA образует анион α по отношению к кислороду и может переходить в 2,3-перегруппировку Виттига. Добавление донорных лигандов, таких как TMEDA и HMPA, может увеличить скорость металлирования и расширить область применения субстрата. Хиральные литийорганические реагенты могут быть доступны посредством асимметричного металлирования.

Направленное орто-металлирование является важным инструментом в синтезе региоспецифических замещенных ароматических соединений. Такой подход к литированию и последующему гашению промежуточных форм лития электрофилом часто лучше, чем электрофильное ароматическое замещение из-за его высокой региоселективности. Эта реакция протекает посредством депротонирования литийорганическими реагентами в положениях α до группы прямого металлирования (DMG) на ароматическом кольце. DMG часто представляет собой функциональную группу, содержащую гетероатом, который является основанием Льюиса и может координироваться с кислотным катионом лития Льюиса. Это создает комплексно-индуцированный эффект близости, который направляет депротонирование в α-положении с образованием частиц ариллития, которые могут далее реагировать с электрофилами. Некоторые из наиболее эффективных DMG - это амиды, карбаматы, сульфоны и сульфонамиды. Это сильные электроноакцепторные группы, которые увеличивают кислотность альфа-протонов на ароматическом кольце. В присутствии двух DMG металлирование часто происходит перпендикулярно более сильной направляющей группе, хотя также наблюдаются смешанные продукты. Ряд гетероциклов, содержащих кислотные протоны, также могут подвергаться орто-металлированию. Однако для бедных электронами гетероциклов обычно используются основания амида лития, такие как LDA, поскольку было обнаружено, что алкиллитий выполняет присоединение к бедным электронами гетероциклам, а не депротонирование. В некоторых комплексах переходный металл-арен, таких как ферроцен, переходный металл притягивает электронную плотность арена, тем самым делая ароматические протоны более кислыми и готовыми к орто-металлированию.
Добавление алкоксида калия к алкиллитию значительно увеличивает основность литийорганических соединений. Наиболее распространенная «супербаза» может быть образована добавлением KOtBu к бутиллитию, часто обозначаемому как реагенты «LiCKOR». Эти «супероснования» являются реагентами с высокой реакционной способностью и часто стереоселективными. В приведенном ниже примере основание LiCKOR генерирует стереоспецифические частицы кротилбороната посредством металлирования и последующего литий-металлоидного обмена.
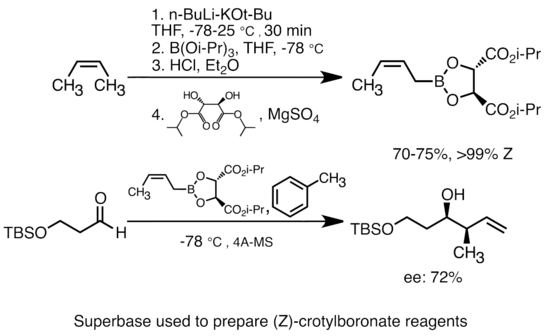
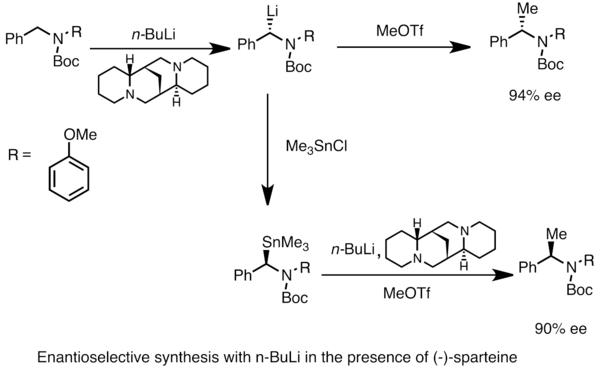
Энантиообогащенные соединения лития могут быть получены посредством асимметричного металлирования прохиральных соединений. субстраты. Асимметричная индукция требует присутствия хирального лиганда, такого как (-) - спартеин. На энантиомерное соотношение хиральных разновидностей лития часто влияют различия в скоростях депротонирования. В приведенном ниже примере обработка N-Boc-N-бензиламина н-бутиллитием в присутствии (-) - спартеина дает один энантиомер продукта с высоким энантиомерным избытком. Трансметаллирование триметилоловохлоридом дает противоположный энантиомер.
еноляты лития образуются в результате депротонирования связи C-H α с карбонильной группой литийорганическими соединениями. Еноляты лития широко используются в качестве нуклеофилов в реакциях образования углерод-углеродных связей, таких как альдольная конденсация и алкилирование. Они также являются важным промежуточным продуктом в образовании силиленольного эфира..

Образование енолята лития можно обобщить как кислотно-основную реакцию, в которой относительно кислый протон α к карбонильной группе (pK = 20-28 в ДМСО) реагирует с литийорганическим основанием. Обычно используются сильные ненуклеофильные основания, особенно амиды лития, такие как LDA, LiHMDS и LiTMP. ТГФ и ДМСО являются обычными растворителями в реакциях енолята лития.
Стереохимия и механизм образования енолята вызвали большой интерес в химическом сообществе. На исход стереохимии енолятов влияют многие факторы, такие как стерические эффекты, растворитель, полярные добавки и типы литийорганических оснований. Среди многих моделей, используемых для объяснения и прогнозирования селективности в стереохимии енолятов лития, есть модель Ирландии.
В этом предположении мономерный LDA реагирует с карбонильным субстратом и образует циклическое переходное состояние типа Циммермана-Тракслера. (E) -енолят является предпочтительным из-за неблагоприятного взаимодействия син-пентана в переходном состоянии (Z) -енолят.

Добавление полярных добавок, таких как HMPA или DMPU, способствует образованию (Z) енолятов. Модель Ирландии утверждает, что эти донорные лиганды координируются с катионами лития, в результате снижается взаимодействие карбонильного кислорода и лития, и переходное состояние не так сильно связано, как шестичленное кресло. Процент (Z) енолятов также увеличивается при использовании оснований лития с более объемными боковыми цепями (таких как LiHMDS). Однако механизм того, как эти добавки изменяют стереоселективность, все еще обсуждается.
Ирландская модель столкнулась с некоторыми трудностями, поскольку она изображает литий как мономер в переходном состоянии. В действительности в растворах енолятов лития часто наблюдаются различные агрегаты лития, и в зависимости от конкретного субстрата, растворителя и условий реакции может быть трудно определить, какой агрегат является действительной реакционноспособной разновидностью в растворе.
Литий-галогенный обмен - это реакция метатезиса между органо-галогенидными и органолитиевыми частицами. Гилман и Виттиг независимо открыли этот метод в конце 1930-х годов.
(2) |
Механизм литий-галогенового обмена все еще обсуждается. Один из возможных путей включает нуклеофильный сложный механизм, который генерирует обратимый« атеизм ». Фарнхам и Калабрезе смогли выделить «ате-комплексный» бис (пентафторфенил) иодинат лития в комплексе с TMEDA и получить кристаллическую структуру рентгеновского излучения. «Ат-комплекс» далее реагирует с электрофилами и дает пентафторфенилйодид и C 6H5Li. Ряд кинетических исследований также подтверждает нуклеофильный путь, по которому карбанион литиевых частиц атакует атом галогена арилгалогенида. Другой возможный механизм включает перенос одного электрона и поколение радикалов. В реакциях вторичного и третичного алкиллития и алкилгалогенидов радикальные частицы были обнаружены с помощью спектроскопии ЭПР. Однако неясно, являются ли эти радикалы промежуточными продуктами реакции. Механистические исследования литий-галогенового обмена также осложняются образованием агрегатов литийорганических соединений.
Скорость обмена галогена лития очень высока. Это обычно быстрее, чем нуклеофильное добавление, и иногда может превышать скорость переноса протона. В приведенном ниже примере обмен между литием и первичным иодидом происходит почти мгновенно и превосходит перенос протона от метанола к трет-бутиллитию. Основной алкеновый продукт образуется с выходом более 90%.

Литий-галогеновый обмен очень полезен при получении новых литийорганических реагентов. Обменные курсы обычно следуют тенденции I>Br>Cl. Алкил- и арилфториды обычно не реагируют с литийорганическими реагентами. Обмен галогенов лития контролируется кинетически, и на скорость обмена в первую очередь влияет стабильность карбанионных промежуточных продуктов (sp>sp2>sp3) литийорганических реагентов. Например, более основные реагенты на основе третичного лития (обычно н-бутиллитий, втор-бутиллитий или трет-бутиллитий) являются наиболее реакционноспособными и будут реагировать с первичным алкилгалогенидом (обычно бромидом или иодидом) с образованием более стабильных литийорганических соединений. Поэтому галогеновый обмен лития наиболее часто используется для получения винил-, арил- и первичных алкиллитиевых реагентов. Обмен галогена лития также облегчается, когда присутствуют алкоксигруппы или гетероатомы для стабилизации карбаниона, и этот метод особенно полезен для приготовления функционализированных литиевых реагентов, которые не могут выдерживать более жестких условий, требуемых для восстановления металлическим литием. Подложки, такие как винилгалогениды, обычно подвергаются литиево-галогеновому обмену с сохранением стереохимии двойной связи.

Ниже приводится пример использования литий-галогенового обмена в синтезе морфина. Здесь н-бутиллитий используется для осуществления литий-галогенового обмена с бромидом. Нуклеофильный карбанионный центр быстро подвергается карболитированию с образованием двойной связи, образуя анион, стабилизированный соседней сульфоновой группой. Внутримолекулярная реакция S N 2 под действием аниона образует циклический каркас морфина.

Обмен галогена лития является важной частью циклизации Пархема. В этой реакции арилгалогенид (обычно йодид или бромид) обменивается на литийорганическое соединение с образованием литиированных аренов. Если арен несет боковую цепь с электрофильным фрагментом, карбанион, присоединенный к литию, будет выполнять внутримолекулярную нуклеофильную атаку и циклизоваться. Эта реакция является полезной стратегией образования гетероцикла. В приведенном ниже примере циклизация по Пархэму использовалась для циклизации изоцианата с образованием изоиндолинона, который затем превращался в нитрон. Нитронные частицы далее вступают в реакцию с радикалами и могут использоваться в качестве «спиновых ловушек» для изучения биологических радикальных процессов.

Литийорганические реагенты часто используются для получения других металлорганических соединений путем трансметаллирования. Медьорганическое соединение, оловоорганическое, кремнийорганическое, борорганическое, фосфорорганическое, церийорганическое и сероорганические соединения часто получают взаимодействием литийорганических реагентов с соответствующими электрофилами.
 | (3) |
Обычные типы трансметаллирования включают обмен Li / Sn, Li / Hg и Li / Te, которые происходят быстро при низкой температуре. Преимущество обмена Li / Sn состоит в том, что предшественники триалкилстаннана подвергаются небольшому количеству побочных реакций, так как в результате образуются n- Побочные продукты Bu 3 Sn не реагируют с алкиллитиевыми реагентами. В следующем примере винилстаннан, полученный гидростаннилированием концевого алкина, образует виниллитий в результате трансметаллирования с помощью n-BuLi.
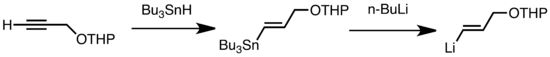
Органолитий. также могут быть использованы для получения цинкорганических соединений путем трансметаллирования солями цинка.

Диорганокупраты лития могут быть образованы путем взаимодействия соединений алкиллития с галогенидом меди (I) е. Получающиеся в результате органокупраты обычно менее реакционноспособны по отношению к альдегидам и кетонам, чем литийорганические реагенты или реагенты Гриньяра.
Наиболее простые алкиллитиевые реагенты и обычные амиды лития коммерчески доступны в различных растворителях и концентрациях. Литийорганические реагенты также можно приготовить в лаборатории. Ниже приведены некоторые распространенные методы приготовления литийорганических реагентов.
Восстановление алкилгалогенида металлическим литием может давать простые алкил- и арилорганические реагенты.
 | (4) |
Промышленное приготовление органолитиевых реагентов достигается с помощью этого метода путем обработки алкилхлорид с металлическим литием, содержащий 0,5-2% натрия. Превращение сильно экзотермическое. Натрий инициирует радикальный путь и увеличивает скорость. Восстановление происходит по радикальному пути. представляет собой пример приготовления функционализированного литиевого реагента с использованием восстановления металлическим литием. Иногда металлический литий в форме тонкодисперсных порошков используется в реакции с некоторыми катализаторами, такими как нафталин или 4,4'- ди-трет-бутилб ифенил (DTBB). Другой субстрат, который может быть восстановлен металлическим литием для образования алкиллитиевых реагентов, - это сульфиды. Восстановление сульфидов полезно при образовании функционализированных литийорганических реагентов, таких как альфа-литиоэфиры, сульфиды и силаны.

Вторым методом получения литийорганических реагентов является металлирование (водородный обмен лития). Относительная кислотность атомов водорода определяет положение литиирования.
Это наиболее распространенный метод приготовления алкиниллитиевых реагентов, поскольку концевой водород, связанный с sp-углеродом, очень кислый и легко депротонируется. Для ароматических соединений положение литирования также определяется управляющим действием групп заместителей. Некоторые из наиболее эффективных управляющих групп заместителей - это алкокси, амидо, сульфоксид, сульфонил. Металлирование часто происходит в орто-положении по отношению к этим заместителям. В гетероароматических соединениях металлирование обычно происходит в орто-положении по отношению к гетероатому.

См. Литий-галогенный обмен (в разделе «Реакционная способность и применение»)
Третий метод получения литийорганического соединения реагенты проходят через галогеновый обмен лития.
трет-бутиллитий или н-бутиллитий являются наиболее часто используемыми реагентами для получения новых литийорганических соединений посредством обмена галогена лития. Литий-галогенный обмен в основном используется для преобразования арил- и алкенилиодидов и бромидов с sp2-атомами углерода в соответствующие литийорганические соединения. Реакция очень быстрая и часто протекает при температуре от -60 до -120 ° C.
Четвертый метод получения литийорганических реагентов - это трансметаллирование. Этот метод можно использовать для получения виниллития.
В реакции Шапиро два эквивалента сильного алкиллитиевого основания реагируют с п-тозилгидразоновыми соединениями с образованием виниллития или при гашении олефинового продукта.
Литийорганические соединения являются высокоактивными веществами и требуют специальных методов обращения. Они часто являются коррозионными, легковоспламеняющимися, а иногда пирофорными (самовозгорание при воздействии кислорода или влаги). Алкиллитиевые реагенты также могут подвергаться термическому разложению с образованием соответствующих алкильных частиц и гидрида лития. Литийорганические реагенты обычно хранят при температуре ниже 10 ° C. Реакции проводят безвоздушными методами. Концентрация алкиллитиевых реагентов часто определяется с помощью титрования.
Литиевые реагенты реагируют, часто медленно, с простыми эфирами, которые, тем не менее, часто используются в качестве растворителей.
| Растворитель | Температура | n-BuLi | s-BuLi | t-BuLi | MeLi | CH2= C (OEt) -Li | CH2= C (SiMe 3) -Li |
|---|---|---|---|---|---|---|---|
| THF | -40 ° C | 338 мин | |||||
| THF | - 20 ° C | 42 мин | |||||
| THF | 0 ° C | 17 ч | |||||
| THF | 20 ° C | 107 мин | >15 часов | 17 часов | |||
| THF | 35 ° C | 10 минут | |||||
| THF/TMEDA | -20 ° C | 55 ч | |||||
| THF/TMEDA | 0 ° C | 340 мин | |||||
| THF/TMEDA | 20 ° C | 40 мин | |||||
| Эфир | -20 ° C | 480 мин | |||||
| Эфир | 0 ° C | 61 мин | |||||
| Эфир | 20 ° C | 153 ч | <30 min | 17 дней | |||
| Эфир | 35 ° C | 31 ч | |||||
| Эфир / TMEDA | 20 ° C | 603 мин | |||||
| DME | -70 ° C | 120 мин | 11 мин | ||||
| DME | -20 ° C | 110 мин | 2 мин | 2 мин | |||
| DME | 0 ° C | 6 мин |