
Войти
| Иммуномодулирующий имидный препарат | |
|---|---|
| Класс препарата | |
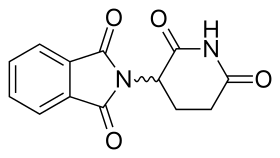 Талидомид Талидомид | |
| Идентификаторы класса | |
| Использовать | узловатая эритема лепрозум, множественная миелома, миелодиспластический синдром, острый миелоидный лейкоз и другие иммунологические состояния |
| код ATC | L04AX |
| Биологическая мишень | TNF, IL-6, VEGF, NF-kB и т. Д. |
| Клинические данные | |
| Drugs.com | Классы лекарств |
| В Викиданных | |
Иммуномодулирующие имидные препараты (IMiD ) - это класс иммуномодулирующих препаратов (препараты, регулирующие иммунные ответы ), содержащий имидную группу. Класс IMiD включает талидомид и его аналоги (леналидомид, помалидомид, ибердомид и апремиласт ). Эти препараты могут также называться «модуляторами цереблона». Цереблон - это белок, на который нацелены препараты этого класса.
Название «IMiD» отсылает как к «IMD» для «иммуномодулирующего препарата», так и к формам имид, имидо-, имид- и имид.
Разработка аналогов талидомида была ускорена открытием антиангиогенных и противовоспалительных свойств лекарство, открывающее новый способ борьбы с раком, а также с некоторыми воспалительными заболеваниями после того, как оно было запрещено в 1961 году. В том числе проблемы с талидомидом; тератогенные побочные эффекты, высокая частота других побочных реакций, плохая растворимость в воде и плохое всасывание из кишечника.
В 1998 г. талидомид был одобрен Управлением по контролю за продуктами и лекарствами США (FDA) для использования при впервые выявленной множественной миеломе (MM) в соответствии со строгими правилами. Это привело к разработке ряда аналогов с меньшим количеством побочных эффектов и повышенной эффективностью, включая леналидомид, помалидомид. и апремиласт, все из которых в настоящее время продаются и производятся Celgene.
Существует три поколения IMiD, каждое последующее поколение лучше переносится и более активно против воспалительных и злокачественных заболеваний.
Талидомид был первоначально выпущен в Федеративной Республике Германии (Западная Германия) под маркой Contergan 1 октября, 1957 год, автор Chemie Grünenthal (ныне Grünenthal ). Препарат в основном назначали как успокаивающее или снотворное, но его также использовали как противорвотное (утреннее недомогание у беременных) и седативное средство. Препарат был запрещен в 1961 году после того, как были обнаружены его тератогенные свойства. Проблемы с талидомидом заключались, помимо тератогенных побочных эффектов, в высокой частоте других побочных реакций наряду с плохой растворимостью в воде и абсорбцией из кишечник. Побочные реакции включают периферическую невропатию у подавляющего большинства пациентов, запор, тромбоэмболию вместе с дерматологическими осложнениями.
Четыре Спустя годы после того, как талидомид был изъят с рынка из-за его способности вызывать серьезные врожденные дефекты, его противовоспалительные свойства были обнаружены, когда пациенты, страдающие от узловатой лепрозной эритемы (ENL), использовали талидомид в качестве седативного средства, и он уменьшил как клинические признаки и симптомы заболевания. В 1991 году было обнаружено, что талидомид ингибирует фактор некроза опухоли-альфа (TNF-α) (5a Sampaio, Sarno, Galilly Cohn and Kaplan, JEM 173 (3) 699-703, 1991). TNF-α представляет собой цитокин, продуцируемый макрофагами иммунной системы, а также медиатор воспалительного ответа. Таким образом, препарат эффективен против некоторых воспалительных заболеваний, таких как ENL (6a Sampaio, Kaplan, Miranda, Nery..... JID 168 (2) 408-414 2008). В 1994 году было обнаружено, что талидомид обладает антиангиогенной и противоопухолевой активностью, что послужило толчком для начала клинических испытаний рака, включая множественную миелому. Обнаружение противовоспалительной, антиангиогенной и противоопухолевой активности талидомида повысило интерес к дальнейшим исследованиям и синтезу более безопасных аналогов.
Леналидомид - первый аналог талидомида, который продается. Он значительно более эффективен, чем его исходное лекарство, только с двумя различиями на молекулярном уровне, с добавленной аминогруппой в положении 4 фталоильного кольца и удалением карбонильной группы из фталоильное кольцо. Разработка леналидомида началась в конце 1990-х годов, а клинические испытания леналидомида начались в 2000 году. В октябре 2001 года леналидомид получил статус орфанных для лечения ММ. В середине 2002 г. он перешел в фазу II, а к началу 2003 г. - в фазу III. В феврале 2003 г. FDA предоставило леналидомиду ускоренный статус для лечения рецидивирующего или рефрактерного ММ. В 2006 г. он был одобрен для лечения ММ вместе с дексаметазоном, а в 2007 г. Европейским агентством по лекарственным средствам (EMA). В 2008 г. в ходе исследования фазы II была выявлена эффективность лечения неходжкинской лимфомы..
Помалидомид (3-аминоталидомид) был вторым аналогом талидомида, поступившим в клинику, будучи более эффективным, чем оба его предшественника. Впервые сообщалось в 2001 году, что помалидомид напрямую ингибирует пролиферацию миеломных клеток и, таким образом, ингибирует ММ как в опухолевых, так и в сосудистых отделах. Эта двойная активность помалидомида делает его более эффективным, чем талидомид, как in vitro, так и in vivo. Этот эффект не связан с ингибированием TNF-α, поскольку сильнодействующие ингибиторы TNF-α, такие как ролипрам и пентоксифиллин, не ингибируют ни рост миеломных клеток, ни ангиогенез. Сообщалось о повышении регуляции интерферона гамма, ИЛ-2 и ИЛ-10 для помалидомида, что может способствовать его антиангиогенной и противомиеломной активности.
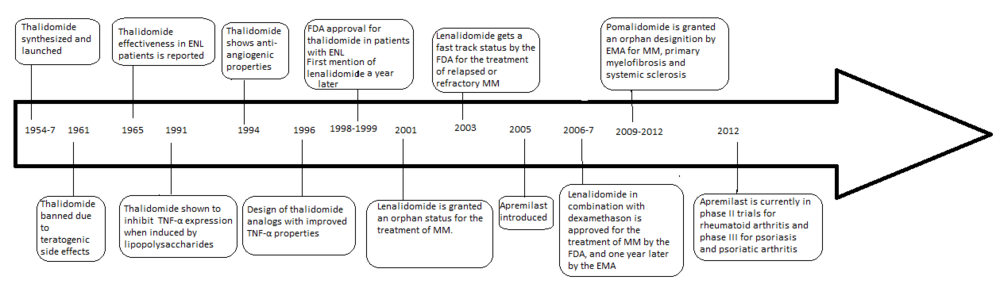 Рисунок 1: Хронологический обзор истории талидомида и его аналогов
Рисунок 1: Хронологический обзор истории талидомида и его аналогов Молекула талидомида является синтетическим производным глутаминовой кислоты и состоит из глутаримида кольцо и фталоильное кольцо (рис. 5). Его IUPAC назывался 2- (2,6-диоксопиперидин-3-ил) изоиндол-1,3-дион, и он имеет один хиральный центр. После сообщения о селективном ингибировании TNF-α талидомидом возобновился Были приложены усилия к клинической разработке талидомида. Клиническая разработка привела к открытию новых аналогов, которые были направлены на улучшение активности и уменьшение побочных эффектов.
Клинически талидомид всегда использовался в качестве рацемата. Обычно изомер S- связан с печально известным тератогенным действием талидомида, а R-изомер лишен тератогенных свойств, но обладает седативным действием, однако эта точка зрения широко обсуждается, и утверждается, что Модель на животных, на которой наблюдались эти различные R- и S-эффекты, не была чувствительна к тератогенным эффектам талидомида. Более поздние сообщения о кроликах, которые являются чувствительным видом, выявили тератогенные эффекты обоих изомеров. Более того, энантиомеры талидомида оказались взаимопревращенными in vivo из-за кислого хирального водорода в асимметричном центре (показано для аналога ЕМ-12 на Фигуре 3), поэтому план введения очищенного одиночный энантиомер во избежание тератогенных эффектов, скорее всего, будет напрасным.
 Рисунок 3: Молекулярная структура ЕМ-12, аналога талидомид. Выделен кислый хиральный водород
Рисунок 3: Молекулярная структура ЕМ-12, аналога талидомид. Выделен кислый хиральный водород Один из представляющих интерес аналогов был получен путем изоиндолиноновой замены фталоильного кольца. Ему было присвоено название ЭМ-12 (рисунок 3). Считалось, что эта замена увеличивает биодоступность вещества из-за повышенной стабильности. Сообщалось, что эта молекула является даже более сильным тератогенным агентом, чем талидомид, для крыс, кроликов и обезьян. Кроме того, эти аналоги являются более сильными ингибиторами ангиогенеза, чем талидомид. Кроме того, амино-талидомид и амино-ЕМ-12 были мощными ингибиторами TNF-α. Эти два аналога позже получили название леналидомид, который является амино аналогом ЕМ-12, и помалидомид, амино аналог талидомида.
 Рисунок 4: Молекулярная структура апремиласта, аналога талидомида
Рисунок 4: Молекулярная структура апремиласта, аналога талидомида После обнаружения нового набора аналогов талидомида, а именно 3- (1,3-диоксо-1,3-дигидроизоиндол-2-ил) -3- (3,4-диметоксифенил) пропионовой кислоты (не показана), которая при наличии активности ингибирования PDE4 началась работа по оптимизации активности. Для этой цели исследователи использовали известную структуру составляющую, 3,4-диалкоксифенил, которая является признанным фармакофором в ингибиторах PDE4, таких как ролипрам (рис. 6). и рофлумиласт и добавил его в структуру ранее упомянутой аналоговой серии. После настройки структуры и тестирования различных замен в положении 4 фталоильного кольца и карбоксильной кислоты исследователи наконец обнаружили молекулу, которая эффективно ингибирует PDE4 и TNF-α, которую они позже назвали апремиластом (рис. 4). Был выбран S-энантиомер апремиласта, поскольку он был более активным энантиомером. Поскольку в структуре апремиласта отсутствует кислотный хиральный водород, он не должен рацемизироваться in vivo, в отличие от талидомида, леналидомида и помалидомида.
.
Основное применение IMiD в медицине - лечение раковые заболевания и аутоиммунные заболевания (включая заболевание, вызванное инфекцией лепрой ). Показания для применения этих агентов, получивших одобрение регулирующих органов, включают:
Показания не по назначению, для которых они кажутся многообещающими, включают:
Талидомид был одобрен FDA для ЭНЛ и ММ в сочетании с дексаметазон. EMA также одобрило его для лечения ММ в сочетании с преднизоном и / или мелфаланом. Признаки для сирот со стороны FDA включают болезнь трансплантат против хозяина, микобактериальную инфекцию, рецидивирующие афтозные язвы, тяжелый рецидивирующий афтозный стоматит, первичные злокачественные новообразования головного мозга, СПИД-ассоциированные синдром истощения, болезнь Крона, саркома Капоши, миелодиспластический синдром и трансплантация гемопоэтических стволовых клеток.
Леналидомид одобрен почти в 70 странах в комбинации с дексаметазоном для лечения пациентов с ММ, которые ранее получали хотя бы одну терапию. Показания для сирот включают диффузную крупноклеточную B-клеточную лимфому, хронический лимфоцитарный лейкоз и лимфому из мантийных клеток. Леналидомид также одобрен для лечения трансфузионно-зависимой анемии из-за миелодиспластических синдромов низкого или промежуточного риска, связанных с цитогенетической аномалией делеции 5q с дополнительными цитогенетическими аномалиями или без них в США, Канаде, Швейцарии, Австралии, New Зеландия, Малайзия, Израиль и несколько стран Латинской Америки, в то время как заявка на регистрацию сбыта в настоящее время проходит оценку в ряде других стран. Многочисленные клинические испытания уже находятся в стадии разработки или проводятся для изучения дальнейшего использования леналидомида, отдельно или в комбинации с другими лекарствами. Некоторые из этих показаний включают острый миелоидный лейкоз, фолликулярную лимфому, MALT-лимфому, макроглобулинемию Вальденстрема, красная волчанка, лимфома Ходжкина, миелодиспластический синдром и др.
Помалидомид был представлен на одобрение FDA 26 апреля 2012 года, а 21 июня было объявлено, что препарат будет получить стандартный обзор FDA. Заявка на регистрацию сбыта была подана в EMA 21 июня 2012 года, где решение могло быть принято уже в начале 2013 года. EMA уже присвоило помалидомиду статус сиротства при первичном миелофиброзе, MM, системном склерозе, пост- полицитемия и пост-эссенциальная тромбоцитемия миелофиброз.
По состоянию на сентябрь 2012 г. апремиласт проходит III фазу испытаний псориаз и исследования фазы II ревматоидного артрита. Эффективность при анкилозирующем спондилите также проходит испытания. По состоянию на март 2014 г. апремиласт одобрен для лечения псориатического артрита. В сентябре 2014 года FDA США одобрило апремиласт для лечения бляшечного псориаза средней и тяжелой степени.
Основными токсическими эффектами одобренных IMiD являются периферическая нейропатия, тромбоцитопения, анемия и венозная тромбоэмболия. Может быть повышенный риск вторичных злокачественных новообразований, особенно острого миелоидного лейкоза у лиц, получающих IMiD.
Тератогенность талидомида является предметом многочисленных дискуссий и лет было выдвинуто множество гипотез. Двумя наиболее известными из них были гипотеза антиангиогенеза и гипотеза модели окислительного стресса со значительными экспериментальными данными, подтверждающими эти две гипотезы относительно тератогенности талидомида.
Недавно появились новые результаты, которые предполагают новый механизм тератогенности.. Цереблон представляет собой белок 51 k Da, локализованный в цитоплазме, ядре и периферической мембране клеток во многих частях тела. Он действует как компонент убиквитинлигазы E3, регулируя различные процессы развития, включая эмбриогенез, канцерогенез и регуляцию клеточного цикла, посредством деградации (убиквитинирования ) неизвестных субстратов. Было показано, что талидомид связывается с церблоном, ингибируя активность убиквитинлигазы E3, что приводит к накоплению субстратов лигазы и подавлению фактора роста фибробластов 8 (FGF8) и FGF10. Это нарушает петлю положительной обратной связи между двумя факторами роста, что может вызвать как множественные врожденные дефекты, так и противомиеломные эффекты.
Результаты также подтверждают гипотезу о том, что увеличение экспрессии церблона является важным элементом противомиеломного эффекта как леналидомида, так и помалидомида. Экспрессия цереблонов была в три раза выше у пациентов, ответивших на лечение, по сравнению с пациентами, не ответившими на лечение, и более высокая экспрессия цереблонов была также связана с частичным или полным ответом, в то время как более низкая экспрессия была связана со стабильным или прогрессирующим заболеванием. 171>Их механизм действия не совсем ясен, но известно, что они ингибируют выработку фактора некроза опухоли, интерлейкина 6 и иммуноглобулина G и VEGF (что приводит к его антиангиогенному действию), костимулирует Т-клетки и NK-клетки и увеличивает гамма-интерферон и производство интерлейкина 2. Их тератогенные эффекты, по-видимому, опосредуются связыванием с церблоном. С другой стороны, апремиласт ингибирует PDE4.
Талидомид и его аналоги, леналидомид и помалидомид, как полагают, действуют аналогичным образом, хотя их точный механизм действия еще полностью не изучен.. Считается, что при различных заболеваниях они действуют через разные механизмы. Чистый эффект, вероятно, связан с сочетанием разных механизмов. Однако считается, что апремиласт действует через другой механизм и поэтому будет обсуждаться отдельно. Механизм действия будет объяснен в свете сегодняшних знаний, в основном, в MM (Рисунок 2).
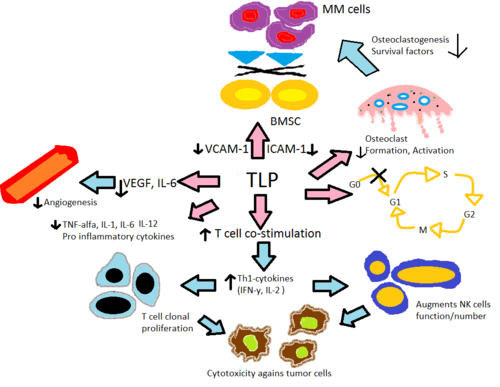 Рис. 2: Механизм TLP при множественной миеломе. TLP относится к талидомиду, леналидомиду и помалидомиду
Рис. 2: Механизм TLP при множественной миеломе. TLP относится к талидомиду, леналидомиду и помалидомиду Талидомид и его иммуномодулирующие аналоги изменяют продукцию воспалительных цитокинов TNF-α, IL-1, ИЛ-6, ИЛ-12 и противовоспалительный цитокин ИЛ-10. Считается, что аналоги ингибируют выработку TNF-α, причем аналоги действуют in vitro в 50 000 раз сильнее, чем исходное лекарственное средство талидомид. Считается, что этот механизм связан с усиленной деградацией TNF-α мРНК, что приводит к уменьшению количества секретируемого провоспалительного цитокина. Это объясняет эффект талидомида при назначении пациентам с ЭНЛ, поскольку у них обычно наблюдается высокий уровень TNF-α в крови и при дерматологических поражениях. Напротив, анализ in vitro продемонстрировал, что TNF-α действительно усиливается при активации Т-клеток, где CD4 + и CD8 + Т-лимфоциты стимулировались анти-CD3, что позже было подтверждено ранние фазы испытаний с участием солидных опухолей и воспалительных дерматологических заболеваний. IL-12 - это еще один цитокин, который подавляется и усиливается талидомидом и его аналогами. Когда моноциты стимулируются липополисахаридами, продукция IL-12 подавляется, но во время стимуляции Т-клеток продукция увеличивается.
Считается, что леналидомид примерно в 1000 раз больше in vitro обладает более сильным противовоспалительным действием, чем талидомид, а помалидомид примерно в 10 раз сильнее, чем леналидомид. Однако стоит отметить, что при сравнении леналидомида и помалидомида клиническая значимость более высокой активности in vitro неясна, поскольку максимальная переносимая доза помалидомида составляет 2 мг в день по сравнению с 25 мг для леналидомида, что приводит к 10-100 в разы более низкая концентрация помалидомида в плазме.
Талидомид и его аналоги помогают костимуляции Т-клеток через комплекс B7 -CD28 путем фосфорилирования тирозин на рецепторе CD28. Данные in vitro предполагают, что эта костимуляция приводит к повышенному высвобождению цитокинов типа Th1 IFN-γ и IL-2, что дополнительно стимулирует пролиферацию клональных Т-клеток и пролиферацию и активность естественных клеток-киллеров. Это усиливает естественную и зависимую от антител клеточную цитотоксичность. Леналидомид и помалидомид примерно в 100-1000 раз более эффективны в стимулировании клональной пролиферации Т-клеток, чем талидомид. Кроме того, данные in vitro предполагают, что помалидомид возвращает Tdiv class="ht" клетки в Th1 за счет усиления фактора транскрипции T-bet.
Ангиогенез или рост новых кровеносных сосудов сообщалось, что это соответствует прогрессированию ММ, где фактор роста эндотелия сосудов (VEGF) и его рецептор, bFGF и IL-6, по-видимому, необходимы для миграции эндотелиальных клеток во время ангиогенеза. Считается, что талидомид и его аналоги подавляют ангиогенез за счет модуляции вышеупомянутых факторов, при этом эффективность антиангиогенной активности леналидомида и помалидомида была в 2-3 раза выше, чем у талидомида в различных анализах in vivo, талидомиде также было показано, что блокирует активность NF-κB посредством блокирования IL-6, и было показано, что NF-κB участвует в ангиогенезе. Ингибирование TNF-α не является механизмом подавления ангиогенеза талидомидом, поскольку многие другие ингибиторы TNF-α не подавляют ангиогенез.
Противоопухолевая активность талидомида in vivo считается, что это связано с мощным антиангиогенным эффектом, а также с изменениями экспрессии цитокинов. Было показано, что анализы in vitro на апоптоз в клетках MM при обработке талидомидом и его аналогами повышают активность каспазы-8. Это вызывает перекрестный обмен сигналами апоптоза между каспазой-8 и каспазой-9, что приводит к косвенной активации активности каспазы-9. Дальнейшая противоопухолевая активность опосредуется посредством ингибирования апоптозного белка-2 и эффектов выживания IGF-1, повышения чувствительности к опосредованной FAS гибели клеток и повышения Связанный с TNF лиганд, индуцирующий апоптоз. Также было показано, что они вызывают дозозависимую G0 /G1 остановку клеточного цикла в клеточных линиях лейкемии, где аналоги показали в 100 раз большую эффективность, чем талидомид.
Роль ангиогенеза в поддержку милеомы была впервые обнаружена Vacca в 1994 году. Они обнаружили, что усиление ангиогенеза костного мозга коррелирует с ростом миеломы, а поддерживающие стромальные клетки являются важным источником ангиогенных молекул в миеломе. Считается, что это главный компонент механизма in vivo, с помощью которого талидомид подавляет множественную миелому.
Кроме того, считается, что воспалительные реакции в костном мозге способствуют развитию многих гематологических заболеваний. Секреция IL-6 костным мозгом стромальными клетками (BMSC) и секреция молекул адгезии VCAM-1, ICAM-1 и LFA, индуцируется в присутствии TNF-α и адгезии клеток MM к BMSC. IL-6 способствует пролиферации клеточных линий MM и ингибированию Fas-опосредованного апоптоза. Талидомид и его аналоги непосредственно снижают повышающую регуляцию IL-6 и опосредованно через TNF-α, тем самым снижая секрецию молекул адгезии, что приводит к меньшему количеству клеток MM, прикрепляющихся к BMSC. Остеокласты становятся высокоактивными во время ММ, что приводит к резорбции кости и секреции различных факторов выживания ММ. Они снижают уровни молекул адгезии, имеющих первостепенное значение для активации остеокластов, уменьшают образование клеток, которые образуют остеокласты, и подавляют катепсин K, важную цистеиновую протеазу, экспрессируемую в остеокласты.
In vitro апремиласт снижает активность PDE4, что приводит к увеличению концентрации циклического аденозинмонофосфата (цАМФ) в иммунной и неиммунные типы клеток, частично ингибирующие выработку многих провоспалительных цитокинов, таких как TNF-α, IFN-γ, IL-2, IL-12 и IL-23 и повышающие выработку анти- воспалительный цитокин ИЛ-10. Сила ингибирования апремиластом продукции TNF-α аналогична леналидомиду.
 Рисунок 5: Талидомид с обозначенной кольцевой системой
Рисунок 5: Талидомид с обозначенной кольцевой системой Поскольку механизм действия талидомида и его аналоги не полностью ясны, и биорецептор этих веществ не был идентифицирован, понимание взаимосвязи между структурой и активностью талидомида и его аналогов в основном получено из молекулярного моделирования и продолжающихся исследований. Информация о SAR талидомида и его аналогов все еще обрабатывается, поэтому любые тенденции, подробно описанные здесь, наблюдаются в ходе отдельных исследований. Исследования в основном были сосредоточены на улучшении ингибирования талидомида TNF-α и PDE4, а также на его антиангиогенезной активности.
Исследования показали, что замена во фталоильном кольце увеличила бы активность ингибирования TNF-α (фиг. 5). Замещение аминогруппы проверяли в различных местах фталоильного кольца (C4, C5, C6, C7) талидомида и ЕМ-12 (описанного ранее). Добавление аминогруппы в месте C4 как на талидомиде, так и на ЕМ-12 привело к гораздо более сильному ингибированию TNF-α. Это также показало, что аминогруппа должна находиться прямо напротив карбонильной группы в изоиндолиноновой кольцевой системе для наиболее сильной активности. Эти аналоги не ингибируют PDE4 и, следовательно, не действуют путем ингибирования PDE4. Другие добавления более длинных и больших групп в положения C4 и C5 фталоильной кольцевой системы талидомида, некоторые с функциональностью олефина, были протестированы с различными результатами. Повышенный ингибирующий эффект по сравнению с талидомидом был отмечен для групп, у которых атом кислорода был присоединен непосредственно к олефину C5 или C4. Добавление йода и брома к C4 или C5 приводило к равной или пониженной активности по сравнению с талидомидом. Эти группы не сравнивались с леналидомидом или помалидомидом.
 Рисунок 6: Ролипрам, выделение 3,4-диалкоксифенильного фрагмента
Рисунок 6: Ролипрам, выделение 3,4-диалкоксифенильного фрагмента  Рисунок 7: Общая структура для ингибирующих PDE4 аналогов талидомида
Рисунок 7: Общая структура для ингибирующих PDE4 аналогов талидомида Общая структура для аналогов, которые ингибируют TNF-α посредством ингибирования PDE4, получают на основе гидролиза глутаримидного кольца талидомида. Эти аналоги не содержат кислого хирального водорода, в отличие от талидомида, и поэтому можно ожидать, что они будут хирально стабильными.
На фенильном кольце 3,4-диалкоксифенильный фрагмент (рис. 6) является известным фармакофором в Ингибиторы PDE4, такие как ролипрам. Оптимальная активность достигается с метоксигруппой в 4-м положении (X2) и большей группой, такой как циклопентокси в 3-м положении углерода (X3). Однако аналоги талидомида, ингибирующие PDE4, не соответствуют SAR аналогов ролипрама. Для аналогов талидомида этоксигруппа в X3 и метоксигруппа в X2, где X1 представляет собой просто водород, давали самое высокое ингибирование PDE4 и TNF-α. Заменители большего размера, чем диэтокси, в положении X2 – X3 имели пониженную активность. Эффекты этих замен, по-видимому, опосредованы стерическими эффектами.
Для положения Y был исследован ряд групп. Замещенные амиды, размер которых превышает размер метиламида (CONHCH 3), снижают активность ингибирования PDE4. Используя карбоновую кислоту в качестве отправной точки, амидная группа имеет аналогичную активность ингибирования PDE4, но обе группы, как было показано, были значительно менее эффективными, чем группа сложного метилового эфира, у которой было примерно шестикратное увеличение ингибирующей активности PDE4. Сульфоновая группа имела такое же ингибирование PDE4, что и группа метилового эфира. Наилучшее ингибирование PDE4 наблюдалось при присоединении нитрильной группы, которая имеет в 32 раза большую ингибирующую активность PDE4, чем карбоксильная кислота. Заместители в Y, приводящие к увеличению ингибирующей активности PDE4, таким образом, следовали в следующем порядке:
Были изучены замещения во фталоильном кольце, и было замечено, что нитрогруппы в месте C4 или C5 снижали активность, но замещение C4 или C5 аминогруппы резко увеличивало ее. Когда было исследовано замещение в положении 4 (Z) на фталоильном кольце, гидроксильные и метоксигруппы, по-видимому, делают аналог менее сильным ингибитором PDE4. Увеличение активности наблюдали с амино и диметиламино в аналогичной степени, но метильная группа улучшала активность больше, чем вышеупомянутые группы. 4-N-ацетиламиногруппа имела несколько более низкую ингибирующую активность в отношении PDE4 по сравнению с метильной группой, но увеличивала ингибирующую активность соединения TNF-α в дополнительной степени. Заместители в Z, ведущие к увеличению ингибирующей активности PDE4, таким образом, следовали в следующем порядке:
 Рисунок 8: Общая структура аналогов талидомида с ингибированием ангиогенеза
Рисунок 8: Общая структура аналогов талидомида с ингибированием ангиогенеза Для активности ингибирования ангиогенеза, по-видимому, требуется интактное глутаримидное кольцо. Различные группы тестировали в положении R. Вещества, которые имели соли азота в качестве группы R, показали хорошую активность. Улучшенная ингибирующая активность ангиогенеза могла быть из-за повышенной растворимости или того, что положительно заряженный азот добавил взаимодействие с активным центром. Тетрафторирование фталоильного кольца, по-видимому, увеличивает ингибирование ангиогенеза.
Ниже приведены схемы для синтез талидомида, леналидомида, помалидомида и апремиласта, как сообщается в известной первичной литературе. Обратите внимание, что эти схемы синтеза не обязательно отражают стратегии органического синтеза, используемые для синтезировать эти отдельные химические соединения.
 Схема 1: Синтез талидомида, более старая процедура
Схема 1: Синтез талидомида, более старая процедура  Схема 2: Новый синтез талидомида, двухстадийная реакция
Схема 2: Новый синтез талидомида, двухстадийная реакция Синтез талидомида обычно проводят, как показано на схеме 1. Этот синтез представляет собой достаточно упрощенный трехэтапный процесс. Обратной стороной этого процесса, однако, является то, что последняя стадия требует высокотемпературной реакции в расплаве, которая требует множественных перекристаллизаций и не соответствует стандартному оборудованию.
Схема 2 представляет собой новый путь синтеза, который был разработан для того, чтобы сделать реакцию более прямой и обеспечить более высокие выходы. В этом способе в качестве исходного материала используется L-глутамин, а не L-глутаминовая кислота, и, позволяя ей реагировать с N-карбэтоксифталимидом, получают N-фталоил- L -глутамин ( 4) с выходом 50–70%. Затем вещество 4 перемешивают в смеси с карбонилдиимидазолом (CDI ) с достаточным количеством 4-диметиламинопиридина (DMAP ) в тетрагидрофуране (ТГФ ) для катализа реакции и нагревают до дефлегмации в течение 15–18 часов. При кипячении из смеси выкристаллизовывается талидомид. На последней стадии достигается выход талидомида 85–93%, в результате чего общий выход составляет 43–63%.
 Схема 3: Синтез помалидомида
Схема 3: Синтез помалидомида Оба аминоаналога получают конденсацией гидрохлорида 3-аминопиперидин-2,6-диона (соединение 3), который синтезируется в двухстадийной реакции из коммерчески доступного Cbz -L-глютамина. Cbz- L -глутамин обрабатывают CDI в кипящем с обратным холодильником THF с получением Cbz-аминоглутаримида. Для удаления Cbz-защитной группы гидрогенолиз, под давлением 50-60 psi водорода с 10% Pd / C, смешанным с этилацетатом и HCl. Полученный гидрохлорид (соединение 3 на схеме 3) затем подвергали взаимодействию с 3-нитрофталевым ангидридом в кипящей уксусной кислоте с получением 4-нитрозамещенного аналога талидомида и нитрогруппы, затем восстанавливали с помощью гидрирования с получением помалидомида. 390>Схема 4: Синтез леналидомида
Леналидомид синтезируется аналогичным способом с использованием соединения 3 (3-аминопиперидин-2,6-дион), обработанного нитрозамещенным метил-2- (бромметил) бензоатом, и гидрирования нитрогруппы.
 Схема 5: Синтез апремиласта
Схема 5: Синтез апремиласта При синтезе апремиласта соединение 3 на схеме 5 получают с выходом 41% из соединения 2, обработанного гексаметилдисилазид лития в смеси с диметилсульфоном лития и эфиратом трифторида бора. Разделение соединения 3 было достигнуто обработкой его N-ацетил- L -лейцином с получением 3S. Затем на последней стадии используется конденсация 3-N-ацетиламинофталевого ангидрида с 3S с получением апремиласта (1S) с выходом 75%.
| Талидомид | ||
|---|---|---|
| Tмакс [препарат] | 4–6 часов у субъектов с MM. | 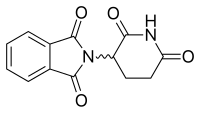 |
| Связывание с белками | 55–65% | |
| Метаболиты | Гидролизованные метаболиты | |
| Период полувыведения [t 1/2 ] | 5,5–7,6 часа | |
| Леналидомид | ||
|---|---|---|
| Tмакс [препарат] | 0,6–1,5 часа у здоровых людей. 0,5–4 часов у субъектов с MM | 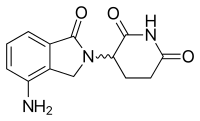 |
| Связывание с белками | ~ 30% | |
| Метаболиты | Еще не изучены | |
| Период полувыведения [t 1/2 ] | 3 часа у здоровых субъектов. 3,1–4,2 часа у субъектов с ММ | |
| Помалидомид | ||
|---|---|---|
| Tмакс [лекарство] | 0,5–8 часов. | 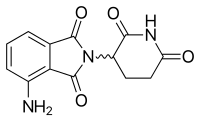 |
| Связывание с белками | Неизвестно | |
| Метаболиты | Неизвестно | |
| Период полувыведения [t 1/2 ] | 6,2–7,9 часа | |
| Апремиласт | ||
|---|---|---|
| Tмакс [препарат] | 1,5–2 часа в здоровые субъекты. В среднем 2 часа у пациентов с тяжелым псориазом бляшечного типа | 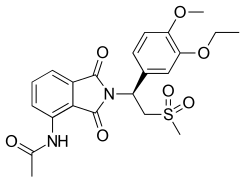 |
| Связывание с белками | ~ 90% | |
| Метаболиты | Глюкуронид O-десметилапремиласта и. деметилированный апремиласт вместе с гидролизованными продуктами | |
| Период полувыведения [t 1/2 ] | 8,2 часов | |