
Войти
 Моделирование ионной жидкости
Моделирование ионной жидкости Молекулярное моделирование включает в себя все методы, теоретические и вычислительные, используемые для моделирования или имитации поведения молекул. Эти методы используются в областях вычислительной химии, дизайна лекарств, вычислительной биологии и материаловедения для изучения молекулярных систем, от небольших химических систем до больших биологических молекул и сборок материалов. Простейшие расчеты можно выполнить вручную, но для молекулярного моделирования любой системы разумного размера неизбежно требуются компьютеры. Общей чертой методов молекулярного моделирования является описание молекулярных систем на атомистическом уровне. Это может включать рассмотрение атомов как наименьшей индивидуальной единицы ( подход молекулярной механики ) или явное моделирование протонов и нейтронов с их кварками, антикварками и глюонами и электронов с их фотонами ( подход квантовой химии ).
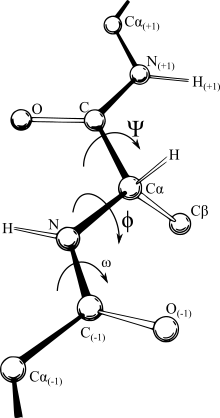 Двугранные углы основной цепи включены в молекулярную модель белка.
Двугранные углы основной цепи включены в молекулярную модель белка. Молекулярная механика - это один из аспектов молекулярного моделирования, поскольку он включает использование классической механики ( механики Ньютона ) для описания физической основы, лежащей в основе моделей. Молекулярные модели обычно описывают атомы (ядро и электроны вместе) как точечные заряды с соответствующей массой. Взаимодействия между соседними атомами описываются пружинными взаимодействиями (представляющими химические связи ) и силами Ван-дер-Ваальса. Для описания последнего обычно используется потенциал Леннарда-Джонса. Электростатические взаимодействия рассчитываются на основе закона Кулона. Атомам назначаются координаты в декартовом пространстве или во внутренних координатах, а также им могут быть присвоены скорости в динамических симуляциях. Скорости атомов связаны с температурой системы, макроскопической величиной. Коллективное математическое выражение называется потенциальной функцией и связано с внутренней энергией системы (U), термодинамической величиной, равной сумме потенциальной и кинетической энергий. Методы, которые минимизируют потенциальную энергию, называются методами минимизации энергии (например, наискорейший спуск и сопряженный градиент ), а методы, которые моделируют поведение системы с течением времени, называются молекулярной динамикой.
Эта функция, называемая потенциальной функцией, вычисляет молекулярную потенциальную энергию как сумму энергетических членов, которые описывают отклонение длин связей, валентных углов и торсионных углов от равновесных значений, плюс члены для несвязанных пар атомов, описывающие ван дер Ваальса и электростатические взаимодействия. Набор параметров, состоящий из длин равновесных связей, валентных углов, значений парциального заряда, силовых констант и ван-дер-ваальсовых параметров, в совокупности называется силовым полем. В различных реализациях молекулярной механики используются разные математические выражения и разные параметры для потенциальной функции. Общие силовые поля, используемые сегодня, были разработаны с использованием химической теории, экспериментальных справочных данных и высокоуровневых квантовых расчетов. Метод, называемый минимизацией энергии, используется для нахождения положений нулевого градиента для всех атомов, другими словами, локального минимума энергии. Состояния с более низкой энергией более стабильны и обычно исследуются из-за их роли в химических и биологических процессах. С другой стороны, моделирование молекулярной динамики вычисляет поведение системы как функцию времени. Он включает в себя решение законов движения Ньютона, в основном второго закона. Интеграция законов движения Ньютона с использованием различных алгоритмов интеграции приводит к атомным траекториям в пространстве и времени. Сила, действующая на атом, определяется как отрицательный градиент функции потенциальной энергии. Метод минимизации энергии полезен для получения статической картины для сравнения состояний аналогичных систем, в то время как молекулярная динамика предоставляет информацию о динамических процессах с внутренним учетом температурных эффектов.
Молекулы можно моделировать либо в вакууме, либо в присутствии растворителя, такого как вода. Моделирование систем в вакууме называется моделированием газовой фазы, в то время как модели, учитывающие присутствие молекул растворителя, называются явным моделированием растворителя. В другом типе моделирования влияние растворителя оценивается с помощью эмпирического математического выражения; это называется моделированием неявной сольватации.
Большинство силовых полей зависят от расстояния, что делает наиболее удобное выражение для этих декартовых координат. Тем не менее, сравнительно жесткий характер связей, которые возникают между конкретными атомами и, по сути, определяют, что подразумевается под обозначением молекулы, делают внутреннюю систему координат наиболее логичным представлением. В некоторых областях представление IC (длина связи, угол между связями и угол закручивания связи, как показано на рисунке) называется представлением Z-матрицы или торсионного угла. К сожалению, непрерывные движения в декартовом пространстве часто требуют прерывистых угловых ветвей во внутренних координатах, что делает относительно трудным работу с силовыми полями во внутренних координатных представлениях, и, наоборот, простое смещение атома в декартовом пространстве может не быть прямой траекторией из-за к запретам взаимосвязанных облигаций. Таким образом, для программ оптимизации вычислений очень часто приходится переключаться между представлениями во время их итераций. Это может доминировать во времени расчета самого потенциала, а в длинноцепочечных молекулах вносит совокупную численную неточность. Хотя все алгоритмы преобразования дают математически идентичные результаты, они различаются по скорости и числовой точности. В настоящее время самым быстрым и точным преобразованием кручения в декартово является метод системы отсчета естественного расширения (NERF).
Методы молекулярного моделирования в настоящее время регулярно используются для исследования структуры, динамики, свойств поверхности и термодинамики неорганических, биологических и полимерных систем. Типы биологической активности, которые были исследованы с помощью молекулярного моделирования, включают фолдинг белка, ферментный катализ, стабильность белка, конформационные изменения, связанные с биомолекулярной функцией, и молекулярное распознавание белков, ДНК и мембранных комплексов.


