
Войти
 |
Дизайн лекарств, часто называемый рациональным дизайном лекарств или просто рациональным дизайном, это изобретательный процесс поиска новых лекарственных средств, основанный на знании биологической мишени. Чаще всего лекарственное средство представляет собой органическую небольшую молекулу, которая активирует или ингибирует функцию биомолекулы, такой как белок, который, в свою очередь, приводит к терапевтической выгоде для пациента. В самом простом смысле, дизайн лекарств включает в себя создание молекул, которые по форме и заряду комплементарны биомолекулярной мишени, с которой они взаимодействуют, и, следовательно, будут связываться с ней. Дизайн лекарств часто, но не обязательно, опирается на методы компьютерного моделирования. Этот тип моделирования иногда называют компьютерным дизайном лекарств . Наконец, дизайн лекарства, основанный на знании трехмерной структуры биомолекулярной мишени, известен как дизайн лекарства на основе структуры . Помимо небольших молекул, биофармацевтические препараты, включая пептиды и особенно терапевтические антитела, становятся все более важным классом лекарств и вычислительных методов для повышения аффинности, селективности и стабильности. этих терапевтических средств на основе белка также были разработаны.
Фраза «разработка лекарственного средства» в некоторой степени является неправильным употреблением. Более точным термином является конструкция лиганда (т.е. конструкция молекулы, которая будет прочно связываться со своей мишенью). Хотя методики проектирования для прогнозирования аффинности связывания достаточно успешны, существует множество других свойств, таких как биодоступность, период полураспада при метаболизме, побочные эффекты и т. Д., это сначала необходимо оптимизировать, прежде чем лиганд станет безопасным и эффективным лекарством. Эти другие характеристики часто трудно предсказать с помощью методов рационального проектирования. Тем не менее, из-за высокого уровня выбытия, особенно во время клинических фаз разработки лекарств, на ранних этапах процесса разработки лекарств больше внимания уделяется выбору лекарств-кандидатов, физико-химические Согласно прогнозам, свойства приведут к меньшему количеству осложнений во время разработки и, следовательно, с большей вероятностью приведут к утвержденному, продаваемому лекарству. Более того, эксперименты in vitro, дополненные вычислительными методами, все чаще используются на ранних этапах открытия лекарств для выбора соединений с более благоприятными ADME (абсорбция, распределение, метаболизм и выведение.) и токсикологические профили.
A биомолекулярная мишень (чаще всего белок или нуклеиновая кислота ) является ключевой молекулой, участвующей в конкретной метаболический или сигнальный путь, связанный с конкретным болезненным состоянием или патологией, или инфекционностью или выживаемостью микроба возбудитель. Потенциальные мишени для лекарств не обязательно вызывают болезнь, но по определению должны быть модифицирующими ее. В некоторых случаях небольшие молекулы будут разработаны для усиления или подавления целевой функции в конкретном пути модификации заболевания. Небольшие молекулы (например, агонисты рецептора , антагонисты, обратные агонисты или модуляторы ; активаторы ферментов или ингибиторы ; или ионные каналы открыватели или блокаторы ), которые будут комплементарны сайту связывания мишени. Небольшие молекулы (лекарства) могут быть сконструированы таким образом, чтобы не воздействовать на другие важные молекулы, не являющиеся мишенями (часто называемые антителами ), поскольку взаимодействие лекарств с молекулами, не являющимися мишенями, может приводить к нежелательным побочные эффекты. Из-за сходства сайтов связывания близкородственные мишени, идентифицированные посредством гомологии последовательностей, имеют наивысший шанс перекрестной реактивности и, следовательно, наибольший потенциал побочных эффектов.
Чаще всего лекарства представляют собой органические небольшие молекулы, полученные путем химического синтеза, но лекарства на основе биополимеров (также известные как биофармацевтические препараты ), производимые с помощью биологические процессы становятся все более распространенными. Кроме того, основанные на мРНК технологии подавления генов могут иметь терапевтическое применение.
В отличие от традиционных методов открытие лекарств (известное как форвардная фармакология ), основанные на пробном и ошибочном испытании химических веществ на культивируемых клетках или животных и сопоставление видимых эффектов с лечением, рациональный дизайн лекарств (также называемый обратной фармакологией ) начинается с гипотезы о том, что модуляция конкретной биологической мишени может иметь терапевтическое значение. Для того, чтобы биомолекула была выбрана в качестве мишени для лекарства, необходимы две важные части информации. Во-первых, это свидетельство того, что модуляция мишени модифицирует болезнь. Эти знания могут быть получены, например, из исследований связи заболеваний, которые показывают связь между мутациями в биологической мишени и определенными болезненными состояниями. Во-вторых, цель - «наркоман ». Это означает, что он способен связываться с небольшой молекулой и что его активность может модулироваться этой небольшой молекулой.
После того, как подходящая мишень идентифицирована, мишень обычно клонируется и произведено и очищено. Затем очищенный белок используют для проведения скринингового анализа. Кроме того, может быть определена трехмерная структура цели.
Поиск небольших молекул, которые связываются с мишенью, начинается со скрининга библиотек потенциальных лекарственных соединений. Это может быть выполнено с помощью скринингового анализа («мокрый экран»). Кроме того, если доступна структура мишени, может быть выполнен виртуальный экран лекарственных препаратов-кандидатов. В идеале лекарственные соединения-кандидаты должны быть «подобными лекарству », то есть они должны обладать свойствами, которые, как ожидается, приведут к пероральной биодоступности, адекватной химической и метаболической стабильности и минимальным токсическим эффектам.. Доступно несколько методов для оценки схожести с лекарственным средством, таких как Правило пяти Липински и ряд методов оценки, таких как липофильная эффективность. В научной литературе также было предложено несколько методов прогнозирования метаболизма лекарств.
Из-за большого количества свойств лекарств, которые необходимо одновременно оптимизировать в процессе разработки, методы многоцелевой оптимизации иногда используются. Наконец, из-за ограничений существующих методов прогнозирования активности разработка лекарств все еще очень зависит от интуитивной интуиции и ограниченной рациональности.
Самая фундаментальная цель в разработке лекарств - предсказать, будет ли данная молекула связываться с мишенью, и если да, то насколько сильно. Молекулярная механика или молекулярная динамика чаще всего используется для оценки силы межмолекулярного взаимодействия между небольшой молекулой и ее биологической мишенью. Эти методы также используются для прогнозирования конформации малой молекулы и моделирования конформационных изменений в мишени, которые могут произойти, когда малая молекула связывается с ней. Полуэмпирические, Неэмпирические методы квантовой химии или теория функционала плотности часто используются для обеспечения оптимальных параметров для расчетов молекулярной механики, а также для оценки электронных свойств (электростатический потенциал, поляризуемость и т.д.) лекарственного средства-кандидата, которое будет влиять на аффинность связывания.,
Методы молекулярной механики также можно использовать для обеспечения полуколичественного предсказания аффинности связывания. Кроме того, основанная на знаниях функция оценки может использоваться для получения оценок сродства связывания. В этих методах используются линейная регрессия, машинное обучение, нейронные сети или другие статистические методы для вывода прогнозных уравнений сродства связывания путем подгонки экспериментальных сродств к полученным с помощью вычислений энергиям взаимодействия между малая молекула и мишень.
В идеале, вычислительный метод должен быть в состоянии предсказать аффинность до того, как соединение будет синтезировано, и, следовательно, теоретически необходимо синтезировать только одно соединение, что значительно экономит время и деньги. Реальность такова, что современные вычислительные методы несовершенны и обеспечивают в лучшем случае только качественно точные оценки аффинности. На практике до того, как будет найдено оптимальное лекарство, все еще требуется несколько итераций дизайна, синтеза и тестирования. Вычислительные методы ускорили открытие, сократив количество требуемых итераций, и часто предоставляют новые структуры.
Разработка лекарств с помощью компьютеров может использоваться на любом из следующих этапов открытия лекарств:
 Блок-схема обычного кластерного анализа для разработки лекарств на основе структуры
Блок-схема обычного кластерного анализа для разработки лекарств на основе структуры Чтобы преодолеть недостаточное предсказание аффинности связывания, рассчитанное с помощью последних функций оценки, для анализа используется информация о взаимодействии белок-лиганд и трехмерная структура соединения. Для разработки лекарств на основе структуры было разработано несколько анализов после скрининга, посвященных взаимодействию белок-лиганд, для улучшения обогащения и эффективного поиска потенциальных кандидатов:
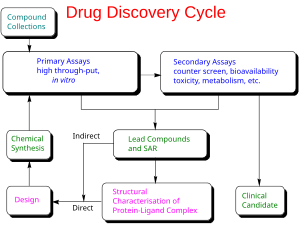
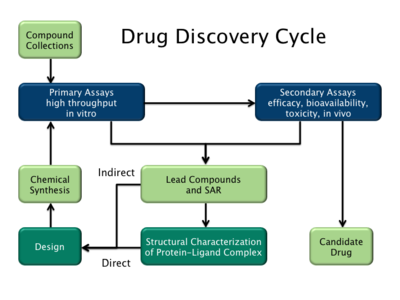 Цикл открытия лекарств, выделяющий как основанные на лигандах (непрямые), так и основанные на структуре (прямые) стратегии создания лекарств.
Цикл открытия лекарств, выделяющий как основанные на лигандах (непрямые), так и основанные на структуре (прямые) стратегии создания лекарств. Существует два основных типа дизайна лекарств. Первый называется дизайн лекарственного средства на основе лиганда, а второй, дизайн препарата на основе структуры .
на основе лиганда Дизайн лекарств (или непрямой дизайн лекарств ) основан на знании других молекул, которые связываются с интересующей биологической мишенью. Эти другие молекулы могут быть использованы для получения модели фармакофора, которая определяет минимально необходимые структурные характеристики, которыми должна обладать молекула для связывания с мишенью. Другими словами, модель биологической мишени может быть построена на основе знания того, что с ней связывается, и эта модель, в свою очередь, может быть использована для конструирования новых молекулярных объектов, которые взаимодействуют с мишенью. В качестве альтернативы количественное соотношение структура-активность (QSAR), в котором может быть получена корреляция между рассчитанными свойствами молекул и их экспериментально определенной биологической активностью. Эти QSAR-отношения, в свою очередь, могут использоваться для прогнозирования активности новых аналогов.
Дизайн лекарственного средства на основе структуры (или прямой дизайн препарата ) опирается на знание трехмерной структуры биологической мишени, полученное с помощью таких методов, как рентгеновская кристаллография или ЯМР-спектроскопия. Если экспериментальная структура мишени недоступна, можно создать модель гомологии мишени на основе экспериментальной структуры родственного белка. Используя структуру биологической мишени, лекарственные препараты-кандидаты, которые, как предполагается, будут связываться с высокой аффинностью и селективностью с мишенью, могут быть разработаны с использованием интерактивной графики и интуиции лекарственного препарата. химик. В качестве альтернативы можно использовать различные автоматизированные вычислительные процедуры, чтобы предложить новые лекарственные препараты-кандидаты.
Современные методы разработки лекарств на основе структуры можно условно разделить на три основные категории. Первый метод - это идентификация новых лигандов для данного рецептора путем поиска в больших базах данных трехмерных структур малых молекул, чтобы найти те, которые подходят для связывающего кармана рецептора, с использованием быстрых приближенных программ стыковки. Этот метод известен как виртуальный скрининг. Вторая категория - это дизайн новых лигандов de novo. В этом методе молекулы лиганда строятся в пределах ограничений связывающего кармана путем пошаговой сборки небольших частей. Эти части могут быть как отдельными атомами, так и фрагментами молекул. Ключевым преимуществом такого метода является то, что можно предложить новые структуры, не содержащиеся в какой-либо базе данных. Третий метод - это оптимизация известных лигандов путем оценки предложенных аналогов в полости для связывания.
Идентификация сайта связывания - это первый шаг в проектировании на основе структуры. Если структура мишени или достаточно похожего гомолога определяется в присутствии связанного лиганда, то лиганд должен наблюдаться в структуре, и в этом случае расположение сайта связывания является тривиальным. Однако могут быть незанятые участки аллостерического связывания, которые могут представлять интерес. Кроме того, возможно, что доступны только структуры апопротеина (белок без лиганда), и надежная идентификация незанятых сайтов, которые могут связывать лиганды с высокой аффинностью, нетривиальна. Вкратце, идентификация сайта связывания обычно основана на идентификации вогнутых поверхностей на белке, которые могут вмещать молекулы размером с лекарство, которые также имеют соответствующие «горячие точки» (гидрофобные поверхности, водород связывания сайтов и т. д.), которые управляют связыванием лигандов.
Дизайн лекарственного средства на основе структуры пытается использовать структуру белков в качестве основы для создания новых лигандов путем применения принципы молекулярного распознавания. Селективное высокое связывание с мишенью обычно желательно, поскольку оно приводит к более эффективным лекарственным средствам с меньшим количеством побочных эффектов. Таким образом, одним из наиболее важных принципов конструирования или получения потенциальных новых лигандов является прогнозирование аффинности связывания определенного лиганда с его мишенью (и известными антителами ) и использование предсказанного сродства в качестве критерия отбора.
Одна ранняя эмпирическая функция оценки общего назначения для описания энергии связывания лигандов с рецепторами была разработана Бёмом. Эта эмпирическая оценочная функция имеет вид:
где:
Более общее термодинамическое «основное» уравнение выглядит следующим образом:
где:
Основная идея заключается в том, что общая свободная энергия связывания может быть разложена на независимые компоненты, которые, как известно, важны для обязательный процесс. Каждый компонент отражает определенный вид изменения свободной энергии во время процесса связывания между лигандом и его рецептором-мишенью. Основное уравнение - это линейная комбинация этих компонентов. Согласно уравнению свободной энергии Гиббса, была построена связь между константой равновесия диссоциации, K d, и компонентами свободной энергии.
Для оценки каждого из компонентов основного уравнения используются различные вычислительные методы. Например, изменение площади полярной поверхности при связывании лиганда можно использовать для оценки энергии десольватации. Количество вращающихся связей, замороженных при связывании лиганда, пропорционально члену движения. Конфигурационная энергия или энергия деформации может быть оценена с использованием расчетов молекулярной механики. Наконец, энергия взаимодействия может быть оценена с использованием таких методов, как изменение неполярной поверхности, статистически выведенные потенциалы средней силы, количество образованных водородных связей и т. Д. На практике компоненты основного уравнения следующие: соответствуют экспериментальным данным с использованием множественной линейной регрессии. Это может быть сделано с помощью разнообразного обучающего набора, включающего множество типов лигандов и рецепторов, чтобы получить менее точную, но более общую «глобальную» модель, или более ограниченный набор лигандов и рецепторов для создания более точной, но менее общей «локальной» модели.
Конкретный пример рационального дизайна лекарств включает использование трехмерной информации о биомолекулах, полученной с помощью таких методов, как рентгеновская кристаллография и ЯМР-спектроскопия. Компьютерный дизайн лекарств, в частности, становится намного более управляемым, когда имеется структура с высоким разрешением целевого белка, связанного с мощным лигандом. Такой подход к открытию лекарств иногда называют дизайном лекарств на основе структуры. Первым однозначным примером применения дизайна лекарственного средства на основе структуры, ведущего к утвержденному лекарству, является ингибитор карбоангидразы дорзоламид, который был одобрен в 1995 году.
Другой Важным примером рационального дизайна лекарственных средств является иматиниб, ингибитор тирозинкиназы, разработанный специально для слитого белка bcr-abl, который характерен для филадельфийской хромосомы -положительного лейкозы (хронический миелогенный лейкоз и иногда острый лимфоцитарный лейкоз ). Иматиниб существенно отличается от предшествующих препаратов для рака, поскольку большинство агентов химиотерапии просто нацелены на быстро делящиеся клетки, не дифференцируя раковые клетки и другие ткани.
Дополнительные примеры. включают:
Утверждалось, что очень жесткий и целенаправленный характер рационального дизайна лекарств подавляет интуитивную в открытии лекарств. Поскольку многие из наиболее важных медицинских открытий были непреднамеренными, недавнее внимание к рациональному дизайну лекарств может ограничить прогресс в открытии лекарств. Кроме того, рациональный дизайн лекарственного средства может быть ограничен грубым или неполным пониманием основных молекулярных процессов заболевания, для лечения которого оно предназначено.

![{\ displaystyle {\ be gin {array} {lll} \ Delta G _ {\ text {bind}} = - RT \ ln K _ {\ text {d}} \\ [1.3ex] K _ {\ text {d}} = {\ dfrac {[ {\ text {Лиганд}}] [{\ text {Receptor}}]} {[{\ text {Complex}}]}} \\ [1.3ex] \ Delta G _ {\ text {bind}} = \ Delta G_ {\ text {desolvation}} + \ Delta G _ {\ text {motion}} + \ Delta G _ {\ text {конфигурация}} + \ Delta G _ {\ text {interface}} \ end {array}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/ba49ddd9dec7415d129787213744ca1afcd2d021)