
Войти
| Фациоскапуло-плечевая мышечная дистрофия | |
|---|---|
| Другие названия | Мышечная дистрофия Ландузи-Дежерина, ФСГМД, ФСГ Воспроизвести медиа |
| Интервал времени экспрессии DUX4 в мышечных клетках FSHD | |
| Специальность | Неврология |
| Симптомы | Слабость лица, крыло лопатки, опущение стопы |
| Обычное начало | Подростковый возраст |
| Продолжение | Долгосрочный |
| Типы | FSHD1, FSHD2 |
| Причины | Генетические (унасанная или новая мутация) |
| Метод диагностики | Генетическое тестирование |
| Дифференциальный диагноз | конечностно-поясная мышечная дистрофия (особенно кальпаинопатия ), болезнь Помпе, митохондриальная миопатия, Полимиозит |
| Ведение | Физиотерапия, фиксация, ортопедическая хирургия |
| Частота | От 1 из 8333 до 1 из 15000 |
Фациоскапуло-плечевая мышечная дистрофчевая чевая чевая ия (FSHD ) - это тип мышечной дистрофии, которая преимущественно ослабляет скел этальные мышцы лица (латинское: facio), те, которые позиционируют лопатку (scapulo), и те, что в плече, лежащая над плечевой костью костью (плечевой). Слабость лопаточных мышц вызывает неправильное расположение лопатки (крылатая лопатка ). В других частях тела, например, в животе и голени, также обычно развивается слабость, что вызывает провисание стопы. Две стороны тела часто поражаются неодинаково. Симптомы обычно начинаются в раннем детстве и становятся заметными в подростковом возрасте, при этом 95% заболевших проявляют болезнь к 20 годам. Немышечные проявления FSHD включают потерю слуха и аномалии кровеносных сосудов в задней части глаза..
FSHD вызваны сложными генетическими изменениями с участием гена DUX4. У тех, у кого нет FSHD, DUX4 экспрессируется (т.е. выключается) в зрелых тканях. При использовании FSHD DUX4 неадекватно распространяется использование ДНК в области DUX4. Эта мутация называется «сокращение D4Z4 » и определяет ЛЛП типа 1 (ЛЛПД1), составляющую 95% случаев ЛЛД. ЛЛПД из-за других мутаций классифицируется как ЛЛП типа 2 (ЛЛПД2). Независимо от того, какая мутация присутствует, заболевание может быть только в том случае, если у человека есть аллель 4qA, который является распространенной вариацией в ДНК рядом с DUX4. До 30% случаев ЛЛПД связаны с новой мутацией, которая может передаваться детям. FSHD1 следует аутосомно-доминантному образцу наследований, что означает, что каждый ребенок аргуутого человека имеет 50% -ный шанс быть аргуутым. Как экспрессия DUX4 повреждение мышц, неясно. Экспрессия гена DUX4 продуцирует белок DUX4, функция которого заключается в модуляции сотен других генов, многие из которых участвуют в функциях мышц. Диагноз ставится на основании генетического тестирования.
Лекарства от ЛЛПД не существует. Никакие фармацевтические препараты не доказали свою эффективность в изменении течения болезни. Симптомы можно устранить с помощью физиотерапии, фиксации и ортопедической хирургии. Хирургическая фиксация лопатки к груд клетке в отдельных случаях уменьшение симптомов со стороны плеча. ЛЛПД является третьим по распространенности генетическим заболеванием скелетных мышц (мышечная дистрофия Дюшенна / мышечная дистрофия Беккера и первой миотонической дистрофией быть вторым), что анализирует от 1 из 8 333 до 1 из 15 000 человек. Прогноз улучшен, многие из них никогда не сталкиваются со значительными ограничениями, хотя до 20% пострадавших людей становятся тяжелыми инвалидами, требующими использования инвалидной коляски или самоката. Ожидаемая продолжительность жизни, как правило, не выделяется, за исключением редких случаев дыхательной недостаточности.
Первым описанием человека с ЛЛПД является отчет о вскрытии 1852 г., хотя ЛЛПД не считалась заболеванием до 1870- х и 1880-х годов, когда французские врачи Ландузи и Дежерин следовали за семьей, пострадавшей от этого; таким образом, ЛЛД иногда называют мышечной дистрофией Ландузи-Дежерина . В 1991 году была установлена связь с верхушкой хромосомы 4, которая, как было обнаружено, связана с сокращением D4Z4 в 1993 году. DUX4 был открыт в 1999 году, но только в 2010 году генетический механизм, вызывающий его экспрессию, был пробнил. В 2012 году была обнаружена преобладающая мутация FSHD2. В 2014 году исследователи опубликовали первое предложенное патофизиологическое определение заболеваний и четыре жизнеспособные терапевтические цели для точек вмешательства.
 Рисунок одного брата в возрасте 17 лет. Видна атрофия плеча. На виде сбоку виден поясничный гиперлордоз. На виде спереди видно истощение большой грудной мышцы.
Рисунок одного брата в возрасте 17 лет. Видна атрофия плеча. На виде сбоку виден поясничный гиперлордоз. На виде спереди видно истощение большой грудной мышцы.  Фотография другого брата в возрасте 21 года. На виде сбоку правая лопатка выглядит вытянутой. На виде сзади правая лопатка выглядит повернутой вниз с боковым смещением.
Фотография другого брата в возрасте 21 года. На виде сбоку правая лопатка выглядит вытянутой. На виде сзади правая лопатка выглядит повернутой вниз с боковым смещением. Мышцы лица, плечевой пояс и плечо классически поражены, хотя эти мышцы могут быть повреждены. сохранились и другие мышцы обычно поражаются. Распределение и степень мышечной слабости различаются даже между однояйцевыми близнецами. Отдельные мышцы могут ослабнуть, в то время как соседние мышцы остаются здоровыми. Слабость мышц обычно становится заметной на одной стороне тела, а не на другой, что является признаком болезни. Мышцы правого плеча поражаются чаще, чем мышцы левого плеча, независимо от рук. Скелетно-мышечная боль очень распространена, чаще всего описывается в шее, плечах, пояснице и части колена. Классически появляются симптомы в возрасте 15–30 лет, хотя также встречаются младенческое начало, несмотря на причинной генетики. Длительные статические фазы, в которых не наблюдается прогрессирования, не редкость. FSHD1 и FSHD2 имеют признаки и симптомы, хотя очень большие делеции D4Z4 в FSHD1 (EcoRI 10-11 kb) более связаны с младенческим началом, прогрессирующей потерей слуха, заболеваниями сетчатки и различными редкими проявлениями. 306>
Слабость обычно начинается в мышцах лица. Обычно поражаются мышцы, окружающие глаза (orbicularis oculi muscle ), что может привести к сну с открытыми веками. Мышца, окружающая рот (orbicularis oris muscle ), также часто поражается, что приводит к неспособности сморщить губы или свистеть. Могут быть трудности с произнесением букв M, B и P или выражением лица, которое выглядит подавленным, злым или усталым. После лицевой слабости обычно развивается слабость в мышцах верхней части туловища, особенно в мышцах, соединяющих плечевой пояс с грудной клеткой (передняя зубчатая мышца и средние и нижние трапециевидные волокна ). Верхние трапеции часто сохраняются. Лопатки становятся повернутыми вниз и вытянутыми, что приводит к крылатой лопатке и наклонным плечам. В запущенных случаях кажется, что лопатка «грыжается» вверх и над грудной клеткой. Распространенная жалоба - трудности при работе с руками над головой. Мышцы вращающейся манжеты обычно сохраняются даже на поздних стадиях заболевания. Другой часто поражающей верхней части туловища является большая грудная мышца, в частности, грудная реберная часть, атрофия которой может выступать горизонтальной передней подмышечной складкой.
После слабости лица и верхней части туловища может «спуститься» на плечи (двуглавая мышца и трехглавая мышца ) и тазовый пояс. Предплечья обычно оставляют без внимания, в результате чего внешний вид можно сравнить с вымышленным персонажем Папай. Иногда наблюдается слабость, которая «пропускает» таз и анализ переднюю большеберцовую мышцу (мышца голени), вызывая провисание стопы. Слабость также может проявляться выпуклостью, поясничным лордозом, невозможностью приседать или неспособностью поворачиваться с одним на другом положении в лежа.. Нижние волокна прямые волокна чаще поражаются, чем верхние волокна в виде положительного признака Бивора. Слабость в ногах может проявляться затруднением ходьбы или легким сгибанием бедер.
Медицинская визуализация (КТ и МРТ) показала повреждение мышц, которое не вызывает очевидных симптомов. Одно исследование МРТ показывает, что обычно поражается большая круглая мышца. Обычно поражается полуперепончатая мышца, часть подколенного сухожилия, причем один автор утверждает, что это «наиболее часто и серьезно поражаемая мышца». Также МРТ показывает, что rectus femoris поражается чаще, чем другие мышцы четырехглавой мышцы, медиальная икроножная мышца поражается чаще, чем латеральная икроножная мышца, и подвздошно-поясничная мышца. мышцы очень часто сохраняются.
Наиболее частым немышечно-скелетным проявлением ЛЛП являются легкие аномалии кровеносных сосудов сетчатки, такие как телеангиэктазии или микроаневризмы, по данным одного исследования заболеваемости на 50%. Эти аномальные кровеносные сосуды обычно не влияют на зрение или здоровье, хотя их тяжелая форма имитирует Коата, состояние обнаруживается примерно в 1% случаев ЛЛПД и чаще связанное с большим делением 4q35. Высокочастотная потеря слуха может вызывать людей с помощью 4q35, но в остальном она не более распространена по с населением в целом. Может быть нарушено дыхание, связанное с кифосколиозом и использование инвалидных колясок; это наблюдается у одной трети пациентов, прикованных к инвалидной коляске. Однако поддержка аппарата искусственной вентиляции легких (ночная или дневная) требуется только в 1% случаев.
Генетика ЛЛПД сложна, что приводит к аномальной экспрессии ген DUX4. У тех, у кого нет FSHD, DUX4 во время экспрессируется эмбриогенезом и в какой-то момент становится репрессированным во всех тканях, кроме семенников. При ЛЛПД происходит неадекватное подавление DUX4, что позволяет эктопически продуцировать белок DUX4 в мышцах, вызывая повреждение мышц. Два генетических элемента необходимы для неадекватной репрессии DUX4. Во-первых, должна быть мутация, которая вызывает гипометилирование ДНК, глобальный DUX4, что делает возможным транскрипцию DUX4 в информационную РНК (мРНК). Несколько мутаций типа гипометилирования, в результате чего FSHD подклассифицируется на FSHD 1 (FSHD1) и FSHD типа 2 (FSHD2).
Второй необходимый генетический элемент - последовательность полиаденилирования ниже по течению от DUX4, который обеспечивает стабильность мРНК DUX4, что позволяет мРНК DUX4 существовать достаточно долго, транслироваться в белок DUX4, вызывая повреждение мускула. В популяции наблюдается по меньшей мере 17 вариаций или морфизмов гаплотипа 4q35 (ДНК, включающая массив повторов D4Z4). Эти 17 вариантов можно условно разделить на группы 4qA и 4qB. Именно аллели 4qA содержат сигналы полиаденилирования, обеспечивающие стабильность мРНК DUX4. Аллели 4qB не имеют последовательностей полиаденилирования.
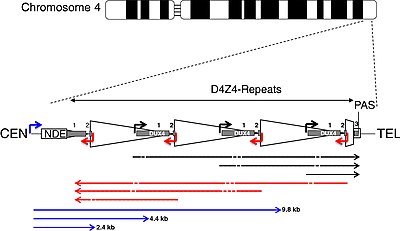 Массив D4Z4 с тремя повторами D4Z4 и аллелем 4qA.
Массив D4Z4 с тремя повторами D4Z4 и аллелем 4qA. | CEN | центррный | TEL | теломерный |
| NDE-бокс | не удаленный элемент | PAS | полиаденилирование сайт |
| треугольник | повтор D4Z4 | трапеция | частичный повтор D4Z4 |
| белый прямоугольник | pLAM | серый прямоугольник | экзоны 1, 2, 3 DUX4 |
| стрелки | |||
| угол | промоторы | прямые | транскрипты РНК |
| черный | смысл | красный | антисмысловой |
| синий | DBE-T | пунктир | сайты нарезки |
DUX4 находится в массиве макросателлитных повторов D4Z4, серии тандемно повторяющихся сегментов ДНК в субтеломерный участок (4q35) хромосомы 4. Каждый повтор D4Z4 имеет длину 3,3 пар оснований (т.п.н.) и является участком эпигенетической регуляции, храним структуры как гетерохроматина, так и эухроматина. При ЛЛД структура гетерохроматина теряется, превращаясь в эухроматин. Название «D4Z4» происходит от устаревшей системы номенклатуры, используемой для сегментов неизвестного значения во время проекта генома человека : Dдля ДНК, 4 для хромосомы 4, Z указывает, что это повторяющаяся последовательность, а 4 - это порядковый номер.
Сообщается, что транскрипты регуляторного элемента D4Z4 (DBT-E) подавляют репрессию DUX4 выражение. Их транскрипция начинается с неудаленного элемента (NDE). Некоторые транскрипты могут деградировать в областях с подобными малыми РНК. DUX4 состоит из трех экзонов. Экзоны 1 и 2 находятся в каждом повторе. Экзон 3 находится в области pLAM, теломерного по отношению к последнему частичному повтору.
FSHD, включающий делецию повторов D4Z4 (так называемое «сокращение D4Z4»), классифицируется как FSHD1, что составляет 95% случаев ЛЛД. Обычно хромосома 4 включает от 11 до 150 повторов D4Z4. В FSHD1 имеется 1–10 повторений D4Z4. Количество повторов примерно обратно коррелирует с тяжестью заболеваний. А именно, те, у кого есть 1-3 повтора, с большей вероятностью будут иметь тяжелое, атипичное и раннее начало заболевания; те, у 4-7 повторов, умеренное заболевание, имеющее большое значение; а у тех, у кого 8-10 повторов, как правило, самые легкие проявления, иногда без симптомов. Сокращение D4Z4 вызывает гипометилирование D4Z4, позволяя транскрипцию DUX4. Удаление всего массива повторов D4Z4 не приводит к FSHD, потому что тогда нет полных копий DUX4 для экспрессии, хотя используются другие врожденные дефекты. Наследование аутосомно-доминантное, хотя 10-30% связаны с de novo (новыми) мутациями.
Субтеломерная область хромосомы 10q содержит перенос тандемных повторов, очень гомологичную (99% идентично) 4q35. Повторы 10q называются «D4Z4-подобными» повторами. Как правило, в случае 10q обычно отсутствует последовательность полиаденилирования, за исключением случая хромосомных перестроек между 4q и 10q, приводящих к сокращению 4q D4Z4 или другого случая переноса повтора 4q D4Z4. и сигнал полиаденилирования на 10кв.
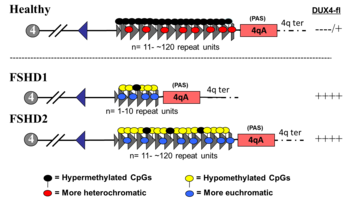 Каждый треугольник представляет собой повтор D4Z4. Кружки над треугольниками обеспечивают метилирование ДНК: высокое метилирование, приводящее к гетерохроматической ДНК, и низкое метилирование, привод к эухроматической ДНК.
Каждый треугольник представляет собой повтор D4Z4. Кружки над треугольниками обеспечивают метилирование ДНК: высокое метилирование, приводящее к гетерохроматической ДНК, и низкое метилирование, привод к эухроматической ДНК. FSHD без сокращений D4Z4 классифицируется как FSHD2, что составляет 5% случаев FSHD. Различные мутации вызывают FSHD2, все приводящие к гипометилированию D4Z4, при котором механизм заболевания сходится с FSHD1. Около 80% случаев FSHD2 происходит из-за деактивирующих мутаций в гене SMCHD1 (структурное поддержание гибкого шарнирного домена хромосом, содержащего 1) на хромосоме 18, гене, ответственном за метилирование ДНК. Дезактивация СМЧД1 приводит к гипометилированию массива повторов D4Z4. Другая причина FSHD2 - мутация в DNMT3B (ДНК-метилтрансфераза 3B), которая также играет роль в метилировании ДНК. По состоянию на 2020 год ранние данные показывают, что третьей причиной FSHD2 является мутация в обеих копиях гена LRIF1, который кодирует белок лиганд-зависимый фактор 1 взаимодействия с ядерным рецептором (LRIF1). Известно, что LRIF1 взаимодействует с белком SMCHD1. По состоянию на 2019 год предположительно существуют дополнительные мутации в других неопознанных генетических местах, которые могут вызывать FSHD2.
Мутация одного аллеля SMCHD1 или DNMT3B может вызвать заболевание. Предварительно было показано, что мутация обоих копий LRIF1 вызывает заболевание у одного человека по состоянию на 2020 год. Как и в случае возникновения FSHD1, для возникновения заболеваний должен присутствовать аллель 4qA. Однако в отличии от формы D4Z4, гены, участвующие в FSHD2, не находятся в непосредственной близости с аллелем 4qA, и поэтому они наследуются независимо от аллеля 4qA, что приводит к дигенному типу наследования. Например, один родитель без FSHD может передать мутацию SMCHD1, а другой родитель, также без FSHD, может передать аллель 4qA, родив ребенка с FSHD2.
Первоначально FSHD1 и FSHD2 были стимулированы как две отдельные генетические причины одного и того же заболевания. Однако их также можно рассматривать не как отдельные причины, как факторы риска. Не редко оба вызывают заболевания у одного и того же человека.
У пациентов с FSHD2, хотя у них нет аллеля 4qA с числом повторов D4Z4 меньше 11, они все же часто имеют один меньше 17 (относительно короткого по сравнению с общей популяцией), предполагая, что большое количество повторов D4Z4 может предотвращает эффекты мутации СМЧД1. Необходимы дальнейшие исследования, чтобы определить верхний предел повторов D4Z4, в которых может встречаться FSHD2.
У с аллелем 4qA и 10 или менее повторами дополнительной мутации SMCHD1, как было показано, показано течение болезни, классифицируя их. как FSHD1, так и FSHD2. У этих людей с FSHD1 / FSHD2 паттерн метилирования массива повторов D4Z4 сходен с таковым у FSHD2. Такое комбинированное представление FSHD1 / FSHD2 наиболее часто встречается у пациентов из 9-10 случаев и редко встречается у пациентов с 8 или менее повторами. Относительное количество мутаций SMCHD1 в группе с 9-10 увеличивает вероятность того, что значительная часть общей популяции имеет 9-10 без, однако при аддитивном эффекте мутации SMCHD1 развиваются симптомы и ставится диагноз. У пациентов с 8 или менее повторами симптомы более вероятны, чем у пациентов с 9-10 повторами, что приводит к диагностике независимо от дополнительной мутации SMCHD1.
Очевидная частота случаев FSHD1 / FSHD2 в 9-10 диапазоне повторов в сочетании с FSHD2-подобным паттерном метилирования предполагает, что размер повторов 9-10 зон перекрытия между FSHD1 и FSDH2.
 Передача сигналов DUX4 в скелетных мышцах, пораженных ЛСД.
Передача сигналов DUX4 в скелетных мышцах, пораженных ЛСД. По состоянию на 2020 год, существует консенсус, что аномальная экспрессия DUX4 в мышцах является причиной ЛЛПД. DUX4 экспрессируется в очень малых количествах, обнаруживаемых в 1 из каждых 1000 незрелых мышечных клеток (миобластов), которые, по-видимому, увеличиваются после созревания миобластов, отчасти, что клетки сливаются по мере созревания, и один Судро, экспрессирующее DUX4, может обеспечивать ядро белок DUX4 соседним ядрам из слитых клеток.
Остается областью активных исследований, как повреждение мышц DUX4. Белок DUX4 - это фактор транскрипции, регулирующий многие другие гены. Некоторые из этих генов участвуют в апоптозе, например, p53, p21, MYC и β-катенин <353.>. Похоже, что DUX4 делает мышечные клетки более склонными к апоптозу, хотя детали механизма до сих пор неизвестны и оспариваются. Другие гены, регулируемые DUX4, участвуют в окислительном стрессе, и кажется, что экспрессия DUX4 снижает устойчивость мышечных клеток к окислительному стрессу. Различия мышц отдельных мышц может частично объяснить паттерны мышц мышцы при ЛЛПД. DUX4 подавляет многие гены, участвуют в развитии мышц, включая MyoD, миогенин, десмин и PAX7. Показано, что DUX4 снижает пролиферацию, дифференцировку и влияние мышечных клеток. Эстроген, по-видимому, играет роль в изменении эффектов DUX4 на дифференцировку мышц, что может объяснить, почему женщины страдают меньше, чем мужчины. DUX4 регулирует несколько генов, которые участвуют в контроле качества РНК, и было показано, что экспрессия DUX4 вызывает накопление РНК с последующим апоптозом.
Ответ клетки гипоксией в одном исследовании сообщалось, что он является основным фактором гибели мышечных клеток, вызванной DUX4. индуцируемые гипоксические факторы (HIF) активируют DUX4, вызывая патологическую передачу сигналов, ведущую к гибели клеток.
Другое исследование показало, чтоия DUX4 в мышечных клетках приводит к привлечению и изменению фиброзные / жировые клетки-предшественники, которые позволяют, почему мышцы заменяются жиром и фиброзной тканью.
Генетическое тестирование является золотым стандартом диагностики ЛЛПД, так как это наиболее чувствительный и специфический доступный тест. Обычно сначала тестируется ЛЛД1. Укороченная длина массива D4Z4 (длина EcoRI от 10 до 38 т.п.н.) со аллелем 4qA поддерживает FSHD1. Если FSHD1 отсутствует, обычно FSHD2 проверяется на предмет следующего, оценивая метилирование в 4q35. Низкое метилирование (менее 20%) в контексте аллеля 4qA достаточно для диагностики. Специфическая мутация, обычно одна из различных мутаций SMCHD1, может быть идентифицирована с помощью секвенирования следующего поколения (NGS).
Измерение длины D4Z4 технически сложно из-за массива повторов D4Z4, состоящего из длинных повторяющихся элементов. Например, NGS бесполезен для оценки длины D4Z4, потому что он разбивает ДНК на фрагменты перед их считыванием, и неясно, из какого повтора D4Z4 произошел каждый секвенированный фрагмент. В 2020 году для массива D4Z4 стало доступно оптическое картирование, которое является более точным и менее трудоемким, чем саузерн-блот. Молекулярное гребнеобразование также доступно для оценки длины массива D4Z4. Иногда 4q или 10q будут иметь комбинацию D4Z4 и D4Z4-подобных повторов из-за обмена ДНК между 4q и 10q, что может дать ошибочные результаты, требующие более детальной обработки.
 Сайты рестрикционных ферментов структуры субтеломерных повторов 4q и 10q
Сайты рестрикционных ферментов структуры субтеломерных повторов 4q и 10q Анализ полиморфизма рестрикционных фрагментов (ПДРФ) был первым разработанным генетическим тестом, который до сих пор используется на 2020 год, хотя постепенно отменяется новыми методами. Он включает разделение ДНК на части рестрикционными ферментами и сортировку полученных рестрикционных фрагментов по размеру с использованием саузерн-блоттинга. Обычно используются рестрикционные ферменты EcoRI и BlnI. EcoRI изолирует массивы повторов 4q и 10q, а BlnI разделяет последовательность 10q на небольшие части, позволяя различать 4q. Фрагмент рестрикции EcoRI состоит из трех частей: 1) проксимальной части 5,7 т.п.н., 2) центрального массива повторов D4Z4 переменного размера и 3) дистальной части, обычно 1,25 т.п.н. Проксимальный участок имеет последовательность ДНК, окрашиваемый зондом p13E-11, который обычно используется для визуализации фрагмента EcoRI во время саузерн-блоттинга. Учитывая, что каждый повтор D4Z4 составляет 3,3 т.п.н., а фрагмент EcoRI содержит 6,9 т.п.н. ДНК, которая представляет собой массив повторов D4Z4, количество D4Z4 может быть рассчитано.
 МРТ, показывающее асимметричное вовлечение различных мышц при ЛЛПД
МРТ, показывающее асимметричное вовлечение различных мышц при ЛЛПД Когда стоимость непомерно высока или диагноз ЛЛПД не подозревается как причина Симптомы Пациенты и врачи могут полагаться на один или несколько из следующих тестов, каждый из которых менее специфичен, чем генетическое тестирование.
Различные проявления лицевой слабости поддаются хирургической коррекции. Золотые имплантаты верхнего века использовались для тех, кто не мог закрыть глаза. Обвисание нижней губы решается с помощью пластической хирургии.

Кинезиологическая лента, накладываемая на лопатки.

Тканевый бандаж для удержания лопаток в втягивании, чтобы уменьшить симптомы со стороны плеча, такие как боль в ключице.

Скапулопексия между лопаткой и лопаткой, до и после операции. Лопатки фиксируютсялантатом ахиллова сухожилия, удерживая их во втянутом положении. На правом изображении хорошо видны большие ромбовидные мышцы.
Распространенность ЛЛПД колеблется от 1 на 8 333 до 1 на 15 000. Нидерланды сообщают о распространенности 1 из 8 333 после учета недиагностированных. Распространенность в США обычно оценивается как 1 на 15 000.
После того, как в 1992 г. стало возможным генетическое тестирование, было обнаружено, что средняя распространенность составляет около 1 на 20 000, что значительно больше, чем до 1992 г. 1 на 20 000 - это, вероятно, заниженная оценка, так как многие с ЛЛПД имеют легкие симптомы и никогда не диагностируются, или они не обращаются за диагнозом.
Не было доказано, что расовая и этническая принадлежность влияет на частоту или тяжесть ЛПД.
Хотя наследование ЛЛПД не показывает склонности к биологическому полу, заболевание реже проявляется у женщин, и даже когда оно проявляется у женщин, они в среднем страдают в меньшей степени, чем поражаются. мужчины. Эстроген считается защитным фактором, который объясняет это несоответствие. Одно исследование показало, что эстроген снижает активность DUX4. Однако другое исследование не обнаружило связи между заболеваниями и пожизненным воздействием эстрогена у женщин. В том же исследовании было обнаружено, что прогрессирование заболевания не отличалось от периодов гормональных изменений, таких как менархе, беременность и менопауза.
Первое описание человека с ЛЛПД в медицинской литературе фигурирует в отчете о вскрытии трупа Жана Крювелье в 1852 году. В 1868 году Дюшенн опубликовал свою основополагающую работу по мышечной дистрофии Дюшенна и как части ее дифференциала было описанием FSHD. Сначала в 1874 г., затем в более цитируемой публикации 1884 г. и снова с фотографиями в 1885 г. французские врачи Луи Ландузи и Жозеф Дежерин опубликовали подробные сведения о болезни, признав ее как отдельная клиническая форма, и поэтому ЛЛПД иногда называют болезнью Ландузи-Дежерина. В своей статье 1886 года Ландузи и Дежерин обратили внимание на семейную природу расстройства и упомянули, что четыре поколения страдают заболеванием. родственники, которых они исследовали. Формального определения клинических особенностей ЛЛПД не было до 1952 года, когда была изучена большая семья из Юты с ЛЛПД. Примерно с 1980 года возрастающий интерес к ЛЛПД привел к более глубокому пониманию большой вариабельности заболевания и растущему пониманию генетических и патофизиологических сложностей. К концу 1990-х исследователи наконец начали понимать области хромосомы 4, связанные с FSHD.
С момента публикации объединяющей теории в 2010 году исследователи продолжали уточнять свое понимание DUX4. С растущим доверием к этой работе исследователи предложили в 2014 году первую консенсусную точку зрения на патофизиологию заболевания и возможные подходы к терапевтическому вмешательству, основанные на этой модели. назывался:
1886
1950
1982
1991
1992
1993
1994
1995
1996
1998
1999
2001
2002
2003
2004
2006
2007
2009
2010
Доктор Фрэнсис Коллинз, который руководил первым секвенированием Генома человека с помощью Национальный институт здравоохранения заявил:
«Если бы мы думали о коллекции величайших хитов ген, это было бы в списке»,
Даниэль Перес, соучредитель, президент и генеральный директор Общество ФСХовало новые открытия, говоря:
«Это долгожданное объяснение точных биологических механизмов болезни, которая поражает примерно одного из 14 000 или 22 100 американцев и 490 000 во всем мире», - сказал он, добавив, это открытие «огромные возможности для исследований по разработке способов предотвращения.
MDA заявило, что:
«Новые результаты облегчат диагностику ЛЛПД у кого-то с симптомами и предсказать, у кого разовьется болезнь в кто-то без симптомов. и инструкциями (РНК ) вызывают проблемы для мышечной ткани при ЛЛПД ».
Цитируется в пресс-релизе Университета Рестера, один из соавторов отчета, Сильвер ван дер Маарель из Университета Лейдена, заявил, что
«Это Удивительно осознавать, что долгое и разочаровывающее путешествие, продолжавшееся почти два десятилетия, теперь завершилось идентификацией единственного небольшой вариант ДНК, который различается у пациентов и людей без заболеваний. Наконец-то у нас есть цель, к которой мы можем приступить ».
2012
2013
2014
Несколько препаратов не показали эффективности.
В 1991 году (до 2019 года именуемое «Общество FSH») было основано двумя людьми с ЛЛПД, Дэниелом Пересом и Стивеном Якобсеном. Общество FSHD собрало финансирование для предоставления грантов для исследований FSHD, выступило за стандартизацию названия болезни как фациоскапуло-плечевую мышечную дистрофию и FSHD, и стало соавтором закона MD-CARE, вступившего в силу. в 2001 г., когда впервые были выделены федеральные ресурсы, в том числе Национальные институты здравоохранения, для лечения всех мышечных дистрофий. Общество FSHD выросло в крупнейшую в мире массовую организацию, выступающую за обучение и медицинские исследования.
В 2009 году FSHD-EUROPE была основана европейскими ассоциациями.
На основе консенсусной модели четыре патофизиологии исследователи осуществляют подход к терапевтическому вмешательству:
Способы измерения болезни важны для оценки эффективности лекарств в клинических испытаниях.
| Классификация | D |
|---|---|
| Внешние ресурсы |