
Войти
Молекулярная графика ( МГ) - это дисциплина и философия изучения молекул и их свойств посредством графического представления. ИЮПАК ограничивает определение представлениями на «устройстве графического отображения». С тех пор как Дальтон «с атомами и Кекула » ы бензол, наблюдается богатая история рукописных атомов и молекул, и эти представления оказали значительное влияние на современных молекулярных графиках. В этой статье основное внимание уделяется использованию компьютеров для создания молекулярной графики. Обратите внимание, однако, что многие программы и системы молекулярной графики имеют тесную связь между графикой и командами редактирования или вычислениями, например, в молекулярном моделировании.
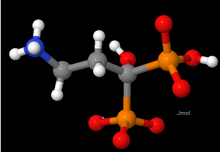 Рис. 1. Обозначения: водород = белый, углерод = серый, азот = синий, кислород = красный и фосфор = оранжевый.
Рис. 1. Обозначения: водород = белый, углерод = серый, азот = синий, кислород = красный и фосфор = оранжевый. Создание молекулярных моделей из физических материалов имеет давнюю традицию. Возможно, наиболее известной является модель ДНК Крика и Ватсона, построенная из стержней и плоских листов, но наиболее широко используемый подход - это представление всех атомов и связей в явном виде с использованием подхода « шар и палка ». Это может продемонстрировать широкий спектр свойств, таких как форма, относительный размер и гибкость. Многие курсы химии предполагают, что студенты будут иметь доступ к моделям мячей и клюшек. Одна из целей основной молекулярной графики состояла в том, чтобы максимально реалистично представить модель «мяч и клюшка» и связать ее с расчетами молекулярных свойств.
На рисунке 1 показана небольшая молекула ( NH 3CH 2CH 2С (ОН) (ПО 3H) (PO 3H) -), нарисованный программой Jmol. Важно понимать, что цвета и формы являются чисто условными, поскольку отдельные атомы не окрашены и не имеют твердых поверхностей. Связи между атомами также не имеют стержневой формы.
Физические модели и компьютерные модели частично дополняют сильные и слабые стороны. Физические модели могут использоваться теми, у кого нет доступа к компьютеру, и теперь их можно дешево изготавливать из пластмассовых материалов. Их тактильные и визуальные аспекты не могут быть легко воспроизведены компьютерами (хотя иногда создаются тактильные устройства). На экране компьютера также трудно оценить гибкость молекул; иллюстрирующее псевдовращение из циклогексана является хорошим примером значения механических моделей.
Однако построить большие физические молекулы сложно, а построение полностью атомных физических моделей даже простых белков может занять недели или месяцы. Более того, физические модели не являются надежными и со временем разрушаются. Молекулярная графика особенно полезна для представления глобальных и локальных свойств молекул, таких как электростатический потенциал. Графика также может быть анимирована для представления молекулярных процессов и химических реакций, что нелегко воспроизвести физически.
Изначально рендеринг выполнялся на экранах ранних электронно-лучевых трубок или с помощью плоттеров, рисовавших на бумаге. Молекулярные структуры всегда были привлекательным выбором для разработки новых инструментов компьютерной графики, поскольку входные данные легко создавать, а результаты обычно очень привлекательны. Первым примером MG была демонстрация белковой молекулы (Project MAC, 1966) Сайрусом Левинталем и Робертом Лэнгриджем. Среди вех в создании высокопроизводительного MG была работа Нельсона Макса по «реалистичному» рендерингу макромолекул с использованием отражающих сфер.
Примерно к 1980 году многие лаборатории как в академических кругах, так и в промышленности признали способность компьютеров анализировать и предсказывать свойства молекул, особенно в материаловедении и фармацевтической промышленности. Дисциплину часто называли «молекулярной графикой», и в 1982 году группа ученых и промышленников в Великобритании основала Общество молекулярной графики (MGS). Первоначально большая часть технологии была сосредоточена либо на высокопроизводительной 3D-графике, включая интерактивное вращение, либо на 3D-рендеринг атомов в виде сфер (иногда с радиацией ). В течение 1980-х годов стал доступен ряд программ для расчета молекулярных свойств (таких как молекулярная динамика и квантовая механика ), и термин «молекулярная графика» часто включал их. В результате MGS теперь изменил свое название на Molecular Graphics and Modeling Society (MGMS).
Требования макромолекулярной кристаллографии также повлияли на MG, потому что традиционные методы построения физических моделей не могли масштабироваться. Первые две белковые структуры, решенные с помощью молекулярной графики без помощи ящика Ричардса, были созданы с помощью программы Стэна Свенсона FIT на графическом дисплее Vector General в лаборатории Эдгара Мейера в Техасском университете Aamp;M: Первая Мардж Легг в лаборатории Эла Коттона в Aamp;M решил вторую структуру стафилококка с более высоким разрешением. нуклеаза (1975), а затем Джим Хогл решил структуру моноклинного лизоцима в 1976 году. Прошел целый год, прежде чем другие графические системы были использованы для замены ящика Ричардса для моделирования плотности в 3-D. Программа FRODO Алвина Джонса (и позже «O») была разработана для наложения молекулярной электронной плотности, определенной с помощью рентгеновской кристаллографии, и гипотетической молекулярной структуры.
В 2009 году BALLView стал первым программным обеспечением, использующим трассировку лучей в реальном времени для молекулярной графики.
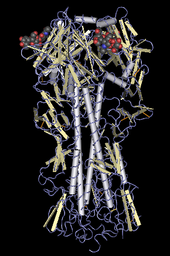 Рис. 2. Изображение гемагглютинина с альфа- спиралями, изображенными в виде цилиндров, а остальная часть цепи - в виде серебряных спиралей. Отдельные белковые молекулы (несколько тысяч) были скрыты. Все неводородные атомы в двух лигандах (предположительно сиаловой кислоты ) показаны в верхней части диаграммы. Обозначения: углерод = серый, кислород = красный, азот = синий.
Рис. 2. Изображение гемагглютинина с альфа- спиралями, изображенными в виде цилиндров, а остальная часть цепи - в виде серебряных спиралей. Отдельные белковые молекулы (несколько тысяч) были скрыты. Все неводородные атомы в двух лигандах (предположительно сиаловой кислоты ) показаны в верхней части диаграммы. Обозначения: углерод = серый, кислород = красный, азот = синий. И компьютерные технологии, и графика внесли свой вклад в молекулярную графику. Развитие структурной биологии в 1950-х годах привело к требованию отображать молекулы с тысячами атомов. Существующие компьютерные технологии были ограничены по мощности, и в любом случае наивное изображение всех атомов оставило зрителей ошеломленным. Поэтому в большинстве систем использовались условные обозначения, в которых информация была неявной или стилистической. Два вектора, встречающиеся в точке, означают атом или (в макромолекулах) полный остаток (10-20 атомов).
Макромолекулярный подход был популяризирован презентацией белков Дикерсоном и Гейсом и графической работой Джейн Ричардсон с помощью высококачественных нарисованных от руки диаграмм, таких как «ленточное» представление. В этом они стремились уловить внутренний «смысл» молекулы. Этот поиск «сообщений в молекуле» всегда сопровождал растущую мощность обработки компьютерной графики. Обычно изображение концентрируется на определенных областях молекулы (таких как активный центр ), и они могут иметь разные цвета или более подробную информацию в количестве явных атомов или типе изображения (например, сферы для атомов).
В некоторых случаях ограничения технологии привели к случайным методам рендеринга. Большинство ранних графических устройств использовали векторную графику, что означало, что рендеринг сфер и поверхностей был невозможен. Программа Майкла Коннолли "MS" рассчитывала точки на доступной для поверхности поверхности молекулы, и эти точки были визуализированы как точки с хорошей видимостью с использованием новой технологии векторной графики, такой как серия Evans and Sutherland PS300. Тонкие срезы («пластины») на структурном дисплее очень ясно показали комплементарность поверхностей для связывания молекул с активными центрами, и «поверхность Коннолли» стала универсальной метафорой.
Связь между искусством и наукой молекулярной графики демонстрируется на выставках, спонсируемых Обществом молекулярной графики. Некоторые экспонаты создаются только с помощью программ молекулярной графики, в то время как другие представляют собой коллажи или включают физические материалы. В примере Майка Ханна (1994), вдохновленного картиной Магритта « Ceci n'est pas une pipe», используется изображение молекулы салметерола. « Ceci n'est pas une Molelele», - пишет Майк Ханн, - «служит для напоминания нам о том, что все графические изображения, представленные здесь, не являются молекулами и даже не изображениями молекул, а изображения значков, которые, как мы полагаем, представляют некоторые аспекты молекулы. характеристики."
Цветная молекулярная графика часто используется на обложках химических журналов в художественной манере.
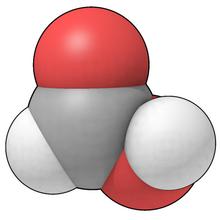 Рис. 4. Модель заполнения пространства муравьиной кислоты. Обозначения: водород = белый, углерод = черный, кислород = красный.
Рис. 4. Модель заполнения пространства муравьиной кислоты. Обозначения: водород = белый, углерод = черный, кислород = красный. Рис. 4 представляет собой "заполняющее пространство" представление муравьиной кислоты, где атомы нарисованы в виде твердых сфер, что указывает на пространство, которое они занимают. Эта и все модели, заполняющие пространство, обязательно являются иконами или абстракциями: атомы - это ядра с электронными «облаками» разной плотности, окружающими их, и как таковые не имеют реальных поверхностей. В течение многих лет размер атомов был аппроксимирован физическими моделями ( CPK ), в которых объемы пластиковых шаров описывают, где находится большая часть электронной плотности (часто размер которых соответствует радиусу Ван-дер-Ваальса ). То есть поверхность этих моделей предназначена для представления определенного уровня плотности электронного облака, а не какой-либо предполагаемой физической поверхности атома.
Поскольку атомные радиусы (например, на рис. 4) лишь немного меньше расстояния между связанными атомами, пиктограммы сферы пересекаются, и в моделях CPK это было достигнуто за счет плоских усечений вдоль направлений связывания, причем сечение было круглым. Когда растровая графика стала доступной, одним из распространенных подходов стало воспроизведение моделей CPK in silico. Относительно просто вычислить круги пересечения, но сложнее представить модель с удалением скрытой поверхности. Полезным побочным продуктом является то, что можно рассчитать обычное значение молекулярного объема.
Сферы часто используются для удобства, поскольку они ограничены как графическими библиотеками, так и дополнительными усилиями, необходимыми для вычисления полной электронной плотности или других величин, заполняющих пространство. Сейчас довольно часто можно увидеть изображения поверхностей, окрашенных для отображения таких величин, как электростатический потенциал. Общие поверхности в молекулярной визуализации включают поверхности, доступные для растворителя («Ли-Ричардс»), поверхности без растворителя («Коннолли») и изоповерхности. Изоповерхность на рис. 5, кажется, показывает электростатический потенциал, при этом синий цвет является отрицательным, а красный / желтый (рядом с металлом) положительным (нет абсолютного соглашения о окраске, а красный / положительный, синий / отрицательный часто меняются местами). Непрозрачные изоповерхности не позволяют увидеть и идентифицировать атомы, и их нелегко вывести. Из-за этого изоповерхности часто рисуются с определенной степенью прозрачности.
Ранние системы интерактивной молекулярной компьютерной графики были машинами векторной графики, в которых использовались векторные мониторы, пишущие штрихами, а иногда даже осциллографы. Электронный луч не перемещается влево и вправо, как на растровом дисплее. Аппаратное обеспечение отображения следовало последовательному списку цифровых инструкций по рисованию (список отображения), непосредственно под углом отрисовывая один штрих для каждой молекулярной связи. Когда список будет завершен, рисование начнется снова с начала списка, поэтому, если список будет длинным (большое количество молекулярных связей), дисплей будет сильно мерцать. Более поздние векторные дисплеи могли вращать сложные структуры с плавным движением, поскольку ориентация всех координат в списке дисплеев могла быть изменена путем загрузки всего нескольких чисел в регистры вращения в дисплейном блоке, и дисплейный блок умножал бы все координаты в отображать список по содержимому этих регистров по мере рисования изображения.
Ранние черно-белые векторные дисплеи могли несколько отличить, например, молекулярную структуру от окружающей ее карты электронной плотности для работы с кристаллографической структурой, нарисовав молекулу ярче, чем карта. Благодаря цветному дисплею их легче отличить. В течение 1970-х годов были доступны двухцветные трубки Penetron с штрих- кодированием, но они не использовались в системах молекулярной компьютерной графики. Примерно в 1980 году компания Evans amp; Sutherland создала первые практические полноцветные векторные дисплеи для молекулярной графики, обычно подключенные к Eamp;S PS-2 или MPS (MPS или Multi-Picture-System относится к нескольким дисплеям, использующим общую стойку графического процессора), графический процессор. Этот ранний цветной дисплей (CSM или Color-Shadow-Mask) был дорогим (около 50 000 долларов), потому что изначально он был спроектирован так, чтобы выдерживать сотрясение движущейся базы авиасимулятора, а также потому, что векторное сканирование управлялось парой (X, Y) усилителей мощностью 1 кВт. Эти системы требовали частого обслуживания, и мудрый пользователь подписал контракт на обслуживание с фиксированной ставкой с Eamp;S. В новых графических процессорах серии Eamp;S PS-300 использовались менее дорогие цветные дисплеи с технологией растрового сканирования, и вся система могла быть куплена дешевле, чем один только старый дисплей CSM.
Цветное растровое графическое отображение молекулярных моделей началось примерно в 1978 году, как показано в этой статье Портера о сферическом затенении атомных моделей. Ранние системы растровой молекулярной графики отображали статические изображения, создание которых могло занять около минуты. Динамически вращающийся цветной растровый молекулярный дисплей постепенно внедрялся в 1982–1985 гг. С появлением программируемого растрового дисплея Ikonas.
Молекулярная графика всегда раздвигала границы технологии отображения и пережила ряд циклов интеграции и разделения вычислительного хоста и дисплея. Ранние системы, такие как Project MAC, были индивидуальными и уникальными, но в 1970-х годах MMS-X и аналогичные системы использовали (относительно) недорогие терминалы, такие как серия Tektronix 4014, часто по коммутируемым линиям на многопользовательские хосты. Устройства могли отображать только статичные картинки, но были способны проповедовать MG. В конце 1970-х отделы (например, кристаллографии) могли позволить себе собственные хосты (например, PDP-11 ) и подключить дисплей (например, PS-1 Evans amp; Sutherland ) непосредственно к автобусу. Список отображения хранился на хосте, и интерактивность была хорошей, поскольку обновления быстро отображались на дисплее - за счет сокращения большинства машин до однопользовательской системы.
В начале 1980-х Evans amp; Sutherland (Eamp;S) отделили свой графический процессор / дисплей PS300, который содержал собственную информацию дисплея, трансформируемую с помощью архитектуры потока данных. Сложные графические объекты могут быть загружены по последовательной линии (например, 9600, 56 Кбод ) или через интерфейс Ethernet, а затем ими можно манипулировать без воздействия на хост. Архитектура превосходна для высокопроизводительных дисплеев, но очень неудобна для расчетов по предметным областям, таких как подгонка электронной плотности и расчеты энергии. Многие кристаллографы и моделисты потратили утомительные месяцы, пытаясь вписать такие действия в эту архитектуру. Компания Eamp;S разработала карту для PS-300, которая имела несколько алгоритмов расчета с использованием конечного автомата шириной 100 бит в попытке упростить этот процесс, но ее было так сложно программировать, что она быстро устарела.
Преимущества для MG были значительными, но к концу 1980-х годов начали появляться рабочие станции UNIX, такие как Sun-3 с растровой графикой (изначально с разрешением 256 на 256). Компьютерный дизайн лекарств, в частности, требовал растровой графики для отображения вычисленных свойств, таких как атомный заряд и электростатический потенциал. Хотя у Eamp;S был высококачественный ассортимент растровой графики (в основном предназначенный для аэрокосмической промышленности), они не смогли отреагировать на вызов рынка низкого уровня, когда рабочие станции покупали отдельные пользователи, а не инженерные подразделения. В результате рынок дисплеев MG перешел к Silicon Graphics вместе с разработкой мини- суперкомпьютеров (например, CONVEX и Alliant ), которые были доступны для хорошо поддерживаемых лабораторий MG. Silicon Graphics предоставила графический язык IrisGL, который был проще в использовании и более производительным, чем архитектура PS300. Коммерческие компании (например, Biosym, Polygen / MSI) перенесли свой код на Silicon Graphics, и к началу 1990-х годов это стало «отраслевым стандартом». Ящики для набора номера часто использовались в качестве устройств управления.
Стереоскопические дисплеи были разработаны на основе жидкокристаллических поляризованных очков, и, хотя на PS2 это было очень дорого, теперь они стали предметом массового потребления. Распространенной альтернативой было добавить поляризуемый экран к передней части дисплея и предоставить зрителям чрезвычайно дешевые очки с ортогональной поляризацией для отдельных глаз. С помощью таких проекторов, как Barco, можно было проецировать стереоскопическое изображение на специальные посеребренные экраны и обеспечивать сотни зрителей очками. Таким образом, молекулярная графика стала широко известна в крупных секторах химической и биохимической науки, особенно в фармацевтической промышленности. Поскольку фон многих дисплеев по умолчанию был черным, на занятиях по моделированию и лекциях было обычным делом проводиться с почти полностью выключенным освещением.
За последнее десятилетие почти вся эта технология превратилась в товар. IrisGL превратился в OpenGL, поэтому молекулярную графику можно запускать на любой машине. В 1992 году Роджер Сэйл опубликовал свою программу РасМол. RasMol содержал очень высокопроизводительный молекулярный рендерер, работавший в Unix / X Window, и Сейл позже перенес его на платформы Windows и Macintosh. Ричардсоны разработали кинемаги и программное обеспечение Mage, которое также было мультиплатформенным. Указав химический тип MIME, можно было бы обслуживать молекулярные модели через Интернет, так что впервые MG можно было распространять с нулевыми затратами независимо от платформы. В 1995 году кафедра кристаллографии Биркбекского колледжа использовала его для проведения «Принципов структуры белка», первого мультимедийного курса в Интернете, в котором приняли участие от 100 до 200 ученых.
 |  |
| Рис. 6. Молекула порина (белка) показана без окклюзии окружающей среды (слева) и с (справа). Расширенные эффекты рендеринга могут улучшить понимание трехмерной формы молекулы. | |
MG продолжает видеть инновации, которые уравновешивают технологию и искусство, и в настоящее время бесплатные программы или программы с открытым исходным кодом, такие как PyMOL и Jmol, получили очень широкое распространение и признание.
В последнее время широкое распространение передового графического оборудования улучшило возможности визуализации инструментов визуализации. Возможности современных языков затенения позволяют включать расширенные графические эффекты (такие как окклюзия окружающей среды, отбрасывание теней и нефотореалистичные методы рендеринга ) в интерактивную визуализацию молекул. Эти графические эффекты, помимо того, что они радуют глаз, могут улучшить понимание трехмерных форм молекул. Пример эффектов, которые могут быть достигнуты с использованием новейшего графического оборудования, можно увидеть в простой системе визуализации с открытым исходным кодом QuteMol.
Рисование молекул требует преобразования между координатами молекул (обычно, но не всегда, в единицах Ангстрема ) и экраном. Поскольку многие молекулы хиральны, важно, чтобы сохранялась правосторонность системы (почти всегда правосторонняя). В молекулярной графике начало координат (0, 0) обычно находится в левом нижнем углу, тогда как во многих компьютерных системах начало координат находится в верхнем левом углу. Если координата z находится за пределами экрана (по направлению к наблюдателю), молекула будет отнесена к правой оси, в то время как изображение на экране будет левым.
Молекулярные превращения обычно требуют:
Конформационные изменения (например, вращение вокруг связей) требуют вращения одной части молекулы относительно другой. Программист должен решить, отражает ли преобразование на экране изменение взгляда или изменение молекулы или ее системы отсчета.
 Рис. 7. Стик-модель кофеина, нарисованная в Jmol.
Рис. 7. Стик-модель кофеина, нарисованная в Jmol. В ранних дисплеях можно было рисовать только векторы (рис. 7), которые легко рисовать, потому что не требуется визуализация или удаление скрытых поверхностей.
На векторных машинах линии были бы гладкими, но на растровых устройствах используется алгоритм Брезенхема (обратите внимание на «неровности» на некоторых связях, которые можно в значительной степени удалить с помощью программного обеспечения сглаживания ).
Атомы можно рисовать в виде кругов, но их следует отсортировать так, чтобы атомы с наибольшими z-координатами (ближайшие к экрану) рисовались последними. Несмотря на несовершенство, это часто дает достаточно привлекательный дисплей. Другие простые приемы, которые не включают алгоритмы скрытых поверхностей:
Типичный псевдокод для создания рис.7 (чтобы молекула точно соответствовала экрану):
// Assume: // Atoms with x, y, z coordinates (Angstrom) and elementSymbol // bonds with pointers/references to atoms at ends // table of colors for elementTypes // find limits of molecule in molecule coordinates as xMin, yMin, xMax, yMax scale = min(xScreenMax / (xMax − xMin), yScreenMax / (yMax − yMin)) xOffset = −xMin × scale yOffset = −yMin × scale for each bond in bonds do atom0 = bond.getAtom(0) atom1 = bond.getAtom(1) x0 = xOffset + atom0.getX() × scale y0 = yOffset + atom0.getY() × scale // (1) x1 = xOffset + atom1.getX() × scale y1 = yOffset + atom1.getY() × scale // (2) x1 = atom1.getX() y1 = atom1.getY() xMid = (x0 + x1) / 2 yMid = (y0 + y1) / 2 color0 = ColorTable.getColor(atom0.getSymbol()) drawLine(color0, x0, y0, xMid, yMid) color1 = ColorTable.getColor(atom1.getSymbol()) drawLine(color1, x1, y1, xMid, yMid)
Обратите внимание, что это предполагает, что начало координат находится в нижнем левом углу экрана, а Y вверху экрана. Многие графические системы имеют начало координат вверху слева, а Y внизу экрана. В этом случае линии (1) и (2) должны иметь генерацию координаты y как:
y0 = yScreenMax -(yOffset + atom0.getY() * scale) // (1) y1 = yScreenMax -(yOffset + atom1.getY() * scale) // (2)
Изменения такого рода изменяют направленность осей, поэтому легко изменить хиральность отображаемой молекулы, если не принять меры.
Для большей реалистичности и лучшего понимания трехмерной структуры молекулы можно использовать многие алгоритмы компьютерной графики. В течение многих лет молекулярная графика подчеркивала возможности графического оборудования и требовала аппаратно-ориентированных подходов. С увеличением мощности компьютеров на настольных компьютерах переносимость становится все более важной, и такие программы, как Jmol, имеют продвинутые алгоритмы, которые не зависят от оборудования. С другой стороны, новейшее графическое оборудование способно интерактивно отображать очень сложные формы молекул с качеством, которое было бы невозможно с помощью стандартных программных методов.
| Разработчики) | Ориентировочная дата | Технология | Комментарии |
|---|---|---|---|
| Кристаллографы | lt;1960 | Нарисованный от руки | Кристаллические структуры со скрытым атомом и удалением связей. Часто клинографические проекции. |
| Джонсон, Мазервелл | ок. 1970 | Перьевой плоттер | ОРТЕП, ПЛУТО. Очень широко применяется для публикации кристаллических структур. |
| Сайрус Левинталь, Боб Лэнгридж, Уорд, Стоц | 1966 г. | Система отображения Project MAC, две степени свободы, джойстик скорости с пружинным возвратом для поворота изображения. | Первое отображение белка на экране. Система для интерактивного построения белковых структур. |
| Барри | 1969 г. | Компьютер LINC 300 с дисплеем осциллографа с двумя кривыми. | Интерактивная система просмотра молекулярной структуры. Ранние примеры динамического вращения, интенсивности глубины метки и параллельного стерео. Раннее использование аппроксимации малых углов (a = sin a, 1 = cos a) для ускорения графических вычислений вращения. |
| Ортони | 1971 г. | Разработал программу просмотра стереозвука (патент Великобритании 13844/70) для молекулярной компьютерной графики. | Горизонтальное двустороннее (наполовину посеребренное) зеркало объединяет изображения, нарисованные на верхней и нижней половинах ЭЛТ. Скрещенные поляризаторы изолируют изображения для каждого глаза. |
| Ортони | 1971 г. | Световое перо, ручка. | Интерактивная система просмотра молекулярной структуры. Выберите бонд, поворачивая другую ручку до тех пор, пока желаемый бонд не загорится по очереди, метод, который позже будет использован в системе MMS-4 ниже, или взяв световым пером. Точки в пространстве указываются с помощью трехмерной «ошибки» под динамическим контролем. |
| Барри, Грэссер, Маршалл | 1971 г. | CHEMAST: компьютер LINC 300, управляющий осциллографом. Двухкоординатный джойстик, аналогичный тому, который позже использовался в ГРИП-75 (см. Ниже). | Интерактивная система просмотра молекулярной структуры. Конструкции динамически вращаются с помощью джойстика. |
| Тунтас и Кац | 1971 г. | Дисплей Adage AGT / 50 | Интерактивная система просмотра молекулярной структуры. Математика вложенного вращения и вращения лабораторного пространства. |
| Перкинс, Пайпер, Таттам, Белый | 1971 г. | Компьютер Honeywell DDP 516, аналоговый компьютер EAL TR48, осциллограф Lanelec, 7 линейных потенциометров. Стерео. | Интерактивная система просмотра молекулярной структуры. |
| Райт | 1972 г. | GRIP-71 на UNC-CH: компьютер IBM System / 360 Model 40 с временным разделением, дисплей IBM 2250, кнопки, световое перо, клавиатура. | Дискретное манипулирование и энергетическая релаксация белковых структур. Программный код стал основой системы ГРИП-75, представленной ниже. |
| Барри и север | 1972 г. | Оксфордский университет: компьютер Ferranti Argus 500, дисплей Ferranti model 30, клавиатура, трекбол, одна ручка. Стерео. | Прототип системы решения кристаллографической структуры больших молекул. Шарик-трекер вращает связь, ручка увеличивает яркость карты молекулярной плотности по сравнению с электронной плотностью. |
| Норт, Форд, Ватсон | Начало 1970-х | Университет Лидса: компьютер DEC PDP 11/40, дисплей Hewlett-Packard. 16 ручек, клавиатура, джойстик с пружинным возвратом. Стерео. | Прототип системы решения кристаллографической структуры больших молекул. Шесть ручек вращают и перемещают небольшую молекулу. |
| Барри, Босхард, Эллис, Маршалл, Фрич, Якоби | 1974 г. | MMS-4: Вашингтонский унив. в Сент-Луисе - компьютер LINC 300 и дисплей LDS-1 / LINC 300, настраиваемые модули отображения. Джойстик вращения, ручки. Стерео. | Прототип системы решения кристаллографической структуры больших молекул. Выберите связку для вращения, поворачивая другую ручку, пока нужная связка не загорится по очереди. |
| Коэн и Фельдманн | 1974 г. | Компьютер DEC PDP-10, дисплей Adage, кнопки, клавиатура, ручки | Прототип системы решения кристаллографической структуры больших молекул. |
| Stellman | 1975 г. | Princeton: компьютер PDP-10, дисплей LDS-1, ручки | Прототип системы решения кристаллографической структуры больших молекул. Карта электронной плотности не показана; вместо этого показатель качества «H-фактор» обновляется по мере изменения молекулярной структуры. |
| Коллинз, Коттон, Хазен, Мейер, Моримото | 1975 г. | CRYSNET, Texas Aamp;M Univ. Компьютер DEC PDP-11/40, дисплей Vector General Series 3, ручки, клавиатура. Стерео. | Прототип системы решения кристаллографической структуры больших молекул. Разнообразие режимов просмотра: покачивание, вращение и несколько режимов отображения стерео. |
| Корнелиус и Краут | 1976 (приблизительно) | Университет Калифорнии в Сан-Диего: эмулятор DEC PDP-11/40 (CalData 135), дисплей Evans and Sutherland Picture System, клавиатура, 6 регуляторов. Стерео. | Прототип системы решения кристаллографической структуры больших молекул. |
| (Йельский университет) | 1976 (приблизительно) | PIGS: компьютер DEC PDP-11/70, дисплей Evans and Sutherland Picture System 2, планшет с данными, ручки. | Прототип системы решения кристаллографической структуры больших молекул. Планшет использовался для большинства взаимодействий. |
| Фельдманн и Портер | 1976 г. | NIH: DEC PDP — 11/70 компьютер. Дисплей Эванса и Сазерленда Picture System 2, ручки. Стерео. | Интерактивная система просмотра молекулярной структуры. Предназначен для интерактивного отображения молекулярных данных из AMSOM - Атласа макромолекулярной структуры на микрофишах. |
| Rosenberger et al. | 1976 г. | MMS-X: Вашингтонский унив. в Сент-Луисе: компьютер TI 980B, дисплей Hewlett-Packard 1321A, видеотерминал Beehive, настраиваемые модули дисплея, пара трехмерных джойстиков с пружинным возвратом, ручки. | Прототип (и позже успешный) система решения кристаллографической структуры больших молекул. Преемник системы MMS-4, описанной выше. Трехмерные джойстики с пружинным возвратом либо перемещают и вращают молекулярную структуру для просмотра, либо молекулярную субструктуру для подгонки, режим управляется тумблером. |
| Бриттон, Липскомб, Пике, Райт, Брукс | 1977 г. | GRIP-75 на UNC-CH: компьютер IBM System / 360 Model 75 с разделением по времени, компьютер DEC PDP 11/45, дисплей Vector General Series 3, трехмерный механизм перемещения от AM Noll и трехмерный джойстик с пружинным возвратом для манипуляций с основанием, Системы измерения вложены джойстик, ручки, ползунки, кнопки, клавиатура, световое перо. | Первое решение кристаллографической структуры больших молекул. |
| Джонс | 1978 г. | FRODO и RING, Институт Макса Планка, Германия, RING: компьютеры DEC PDP-11/40 и Siemens 4004, дисплей Vector General 3404, 6 регуляторов. | Решение кристаллографической структуры крупных молекул. FRODO мог работать на DEC VAX-780 как продолжение RING. |
| Алмазный | 1978 г. | Билдер Кембридж, Англия, компьютер DEC PDP-11/50, дисплей Evans and Sutherland Picture System, планшет. | Решение кристаллографической структуры крупных молекул. Все данные вводятся с помощью планшета. Молекулярные структуры, построенные в режиме онлайн с идеальной геометрией. Позже проходит натяжка с идеализацией. |
| Лэнгридж, Уайт, Маршалл | Конец 1970-х | Ведомственные системы ( PDP-11, дисплеи Tektronix или DEC-VT11, например MMS-X) | Смесь массовых вычислений с ранними дисплеями. |
| Дэвис, Хаббард | Середина 1980-х | ХИМ-Х, ГИДРА | Лабораторные системы с многоцветными, растровыми и векторными приборами (Sigmex, PS300). |
| Биосим, Трипос, Полиген | Середина 1980-х | Устойчивые терминалы PS300 и более дешевые (VT200, SIGMEX) | Коммерческие интегрированные пакеты моделирования и отображения. |
| Силиконовая Графика, Солнце | Конец 1980-х | Рабочие станции IRIS GL (UNIX) | Однопользовательские рабочие станции со стереоскопическим дисплеем по стандартной цене. |
| EMBL - ЧТО ЕСЛИ | 1989, 2000 | Независимый от машины | Почти бесплатный, многофункциональный, все еще полностью поддерживаемый, на нем много бесплатных серверов |
| Сэйл, Ричардсон | 1992, 1993 | RasMol, Kinemage | Платформенно-независимый MG. |
| MDL (ван Влит, Маффет, Адлер, Холт) | 1995–1998 | Перезвон | проприетарный C ++; бесплатный плагин для браузера Mac (OS9) и ПК |
| МолСофт | 1997- | ICM-браузер | проприетарный; бесплатно скачать для Windows, Mac и Linux. |
| 1998- | MarvinSketch и MarvinView. MarvinSpace (2005) | проприетарный Java- апплет или отдельное приложение. | |
| Усилия сообщества | 2000- | DINO, Jmol, PyMol, Avogadro, PDB, OpenStructure | Апплет Java с открытым исходным кодом или отдельное приложение. |
| НОЧ | 2002- | NOC | Исследователь молекулярной структуры с открытым исходным кодом |
| LION Bioscience / EMBL | 2004- | SRS 3D | Бесплатная система с открытым исходным кодом на основе Java3D. Интегрирует трехмерные структуры с данными о последовательностях и функциях (домены, SNP и т. Д.). |
| Суперкомпьютерный центр Сан-Диего | 2006- | Сириус | Бесплатно для академических / некоммерческих организаций |
| Усилия сообщества | 2009- | Программы просмотра HTML5 / JavaScript (веб-компоненты ChemDoodle, GLMol, jolecule, pv, Molmil, iCn3D, 3DMol, NGL, Speck, xtal.js, UglyMol, LiteMol, JSmol) | Все с открытым исходным кодом. Требовать поддержки WebGL в браузере (кроме JSmol). |
До того, как можно было использовать компьютерную графику, примерно в 1976-1977 годах (ссылки и объяснения в нескольких абзацах ниже), использовались механические методы, чтобы подогнать большие молекулы к их картам электронной плотности. Используя методы рентгеновской кристаллографии, кристалл вещества бомбардировали рентгеновскими лучами, и дифрагированные лучи, которые выходили, собирались с помощью компьютера с помощью преобразования Фурье в обычно размытое трехмерное изображение молекулы, видимое путем рисования контура. кружки вокруг высокой электронной плотности для построения контурной карты электронной плотности.
Раньше контурные карты электронной плотности рисовали вручную на больших пластиковых листах. Иногда фишки для бинго помещали на пластиковые листы, где интерпретировались атомы.
Он был заменен коробкой Ричардса, в которой регулируемая латунная молекулярная модель Кендрю была помещена перед двусторонним зеркалом, за которым были пластиковые листы карты электронной плотности. Это оптически наложило молекулярную модель и карту электронной плотности. Модель была перемещена в пределах контурных линий наложенной карты. Затем координаты атомов записывались с помощью отвеса и метра. Компьютерная графика давала надежду значительно ускорить этот процесс, а также дать более четкое представление, потому что небольшая интересующая область могла быть рассмотрена, не заслоняя беспорядок от остальной контурной молекулы, могла быть очерчена ортогональными кольцами электронной плотности. вместо колец в одной плоскости, что дает более однородный обзор облаков, а интересующая область может контролироваться с помощью джойстика с любого направления, а не только с направления обзора через стеклянную панель ящика Ричарда.
Заслуживающая внимания попытка преодолеть низкую скорость графических дисплеев начала 1970-х годов была предпринята в Вашингтонском университете в Сент-Луисе, США. Группа Дэйва Барри попыталась перескочить через современный уровень в области графических дисплеев, создав специальное аппаратное обеспечение для отображения изображений, достаточно сложных для решения кристаллографической структуры крупных молекул, подгоняя молекулы к их картам электронной плотности. Модули дисплея MMS-4 (таблица выше) были медленными и дорогими, поэтому для системы MMS-X (таблица выше) было произведено второе поколение модулей.
Первой крупной молекулой, атомная структура которой была частично определена с помощью системы молекулярной компьютерной графики, была передаточная РНК, созданная командой Сунг-Хоу Кима в 1976 году после первоначальной установки на механический бокс Ричардса. Первая большая молекула которого атомной структура была полностью определена на молекулярном системе компьютерной графики называется нейротоксин А из яда морской змеи, Филиппин по Tsernoglou, Petsko, и Тем, с утверждением того, чтобы быть первым в 1977 году Ричардсон группы опубликовали результаты по частичной атомной структуре протеин-супероксиддисмутазы в том же 1977 году. Все они были сделаны с использованием системы GRIP-75 (таблица выше).
Другие системы подгонки структуры, FRODO, RING, Builder, MMS-X и т. Д. (Таблица выше) также преуспели в решении больших белковых структур в 1977-1980 годах.
Причина, по которой большинство этих систем добились успеха именно в те годы, 1976-1980 гг., Не раньше и не позже, а в короткий промежуток времени, была связана с появлением коммерческого оборудования, которое было достаточно мощным. Две вещи были необходимы и пришли примерно в одно и то же время. Во-первых, карты электронной плотности велики и требуют либо компьютера с адресным пространством не менее 24 бит, либо комбинации компьютеров с меньшим 16-битным адресным пространством плюс несколько лет для преодоления трудностей адресного пространства размером меньше данные. Вторым появлением стали интерактивные компьютерные графические дисплеи, которые были достаточно быстрыми для отображения карт электронной плотности, контурные круги которых требуют отображения множества коротких векторов. Первыми такими дисплеями были Vector General Series 3 и Evans and Sutherland Picture System 2, MultiPicture System и PS-300.
Позже подгонка молекулярной структуры к карте электронной плотности была в значительной степени автоматизирована с помощью алгоритмов с компьютерной графикой в качестве руководства к процессу. Примерами являются программы XtalView и XFit.