
Войти
 Пример молекулярно-динамического моделирования в простой системе: осаждение одного атома меди (Cu) на холодный кристалл меди ( поверхность индекса Миллера (001)). Каждый кружок представляет положение одного атома. Кинетическая энергия атома, приближающегося сверху, перераспределяется между другими атомами, поэтому вместо того, чтобы оттолкнуться, он остается прикрепленным из-за сил притяжения между атомами.
Пример молекулярно-динамического моделирования в простой системе: осаждение одного атома меди (Cu) на холодный кристалл меди ( поверхность индекса Миллера (001)). Каждый кружок представляет положение одного атома. Кинетическая энергия атома, приближающегося сверху, перераспределяется между другими атомами, поэтому вместо того, чтобы оттолкнуться, он остается прикрепленным из-за сил притяжения между атомами.  Моделирование молекулярной динамики часто используется для изучения биофизических систем. Здесь изображена симуляция воды за 100 пс.
Моделирование молекулярной динамики часто используется для изучения биофизических систем. Здесь изображена симуляция воды за 100 пс. 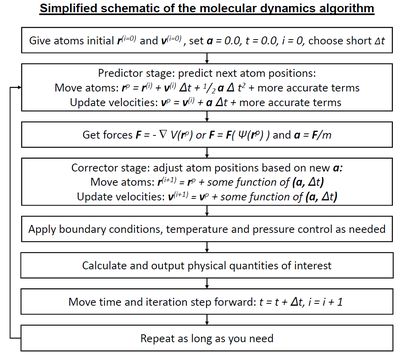 Упрощенное описание стандартного алгоритма моделирования молекулярной динамики, когда используется интегратор типа предиктор-корректор. Силы могут исходить либо от классических межатомных потенциалов (математически описываемых как), либо квантово-механических (математически описываемых как) методов. Между разными интеграторами существуют большие различия; некоторые не имеют точно таких же членов высшего порядка, как указано в блок-схеме, многие также используют производные по времени более высокого порядка, а некоторые используют как текущий, так и предыдущий временной шаг в схемах шагов с переменным временем.
Упрощенное описание стандартного алгоритма моделирования молекулярной динамики, когда используется интегратор типа предиктор-корректор. Силы могут исходить либо от классических межатомных потенциалов (математически описываемых как), либо квантово-механических (математически описываемых как) методов. Между разными интеграторами существуют большие различия; некоторые не имеют точно таких же членов высшего порядка, как указано в блок-схеме, многие также используют производные по времени более высокого порядка, а некоторые используют как текущий, так и предыдущий временной шаг в схемах шагов с переменным временем. Молекулярная динамика ( МД) является компьютерным моделированием метода анализа физических движений из атомов и молекул. Атомам и молекулам позволяют взаимодействовать в течение фиксированного периода времени, что дает представление о динамической «эволюции» системы. В наиболее распространенной версии траектории атомов и молекул определяются путем численного решения уравнений движения Ньютона для системы взаимодействующих частиц, где силы между частицами и их потенциальные энергии часто вычисляются с использованием межатомных потенциалов или силовых полей молекулярной механики. Метод применяется в основном в химической физике, материаловедении и биофизике.
Поскольку молекулярные системы обычно состоят из огромного числа частиц, невозможно определить свойства таких сложных систем аналитически; МД-моделирование позволяет обойти эту проблему с помощью численных методов. Однако длительное моделирование MD математически плохо обусловлено, порождая кумулятивные ошибки в численном интегрировании, которые можно минимизировать с помощью правильного выбора алгоритмов и параметров, но не устранить полностью.
Для систем, которые подчиняются эргодической гипотезе, эволюция одного моделирования молекулярной динамики может использоваться для определения макроскопических термодинамических свойств системы: средние по времени эргодической системы соответствуют средним микроканоническим ансамблям. MD также называют "статистической механикой в числах" и "видением Лапласа ньютоновской механики " предсказания будущего путем оживления сил природы и предоставления возможности проникновения в суть молекулярного движения в атомном масштабе.
Изначально MD был разработан в начале 1950-х годов после более ранних успехов моделирования методом Монте-Карло, которые, в свою очередь, восходят к восемнадцатому веку, например, в задаче об игле Бюффона, но популяризировали статистическую механику в Национальной лаборатории Лос-Аламоса Розенблютом и Метрополисом. в том, что сегодня известно как алгоритм Метрополиса – Гастингса. Интерес к эволюции систем N-тел во времени возник гораздо раньше, в семнадцатом веке, начиная с Ньютона, и продолжился в следующем столетии, в основном с акцентом на небесную механику и такие вопросы, как стабильность Солнечной системы. Многие численные методы, используемые сегодня, были разработаны в этот период времени, который предшествовал использованию компьютеров; например, наиболее распространенный алгоритм интеграции, используемый сегодня, алгоритм интеграции Верле, был использован еще в 1791 году Жаном Батистом Жозефом Деламбром. Численные расчеты с использованием этих алгоритмов можно рассматривать как МД «вручную».
Еще в 1941 г. интегрирование уравнений движения многих тел производилось с помощью аналоговых компьютеров. Некоторые предприняли трудоемкую работу по моделированию движения атомов путем построения физических моделей, например, с использованием макроскопических сфер. Цель состояла в том, чтобы расположить их таким образом, чтобы воспроизвести структуру жидкости и использовать ее для изучения ее поведения. Дж. Д. Бернал сказал в 1962 году: «. .. Я взял несколько резиновых мячей и склеил их стержнями разной длины от 2,75 до 4 дюймов. Я попытался сделать это в первую очередь как можно небрежнее., работаю в собственном офисе, меня перебивают каждые пять минут или около того, и я не помню, что делал до перерыва ".
После открытия микроскопических частиц и разработки компьютеров интерес расширился за пределы полигона гравитационных систем к статистическим свойствам материи. В попытке понять происхождение необратимости Ферми предложил в 1953 году и опубликовал в 1955 году использование MANIAC I, также в Национальной лаборатории Лос-Аламоса, для решения временной эволюции уравнений движения для объекта системы многих тел. к нескольким вариантам законов силы; сегодня эта основополагающая работа известна как проблема Ферми – Паста – Улама – Цинго. Изменение энергии исходной работы во времени показано на рисунке справа.
 Одно из самых первых симуляций системы N-тел было выполнено на MANIAC-I Ферми и его коллегами, чтобы понять происхождение необратимости в природе. Здесь показана зависимость энергии от времени для системы из 64 частиц.
Одно из самых первых симуляций системы N-тел было выполнено на MANIAC-I Ферми и его коллегами, чтобы понять происхождение необратимости в природе. Здесь показана зависимость энергии от времени для системы из 64 частиц. В 1957 году Олдер и Уэйнрайт использовали компьютер IBM 704 для моделирования совершенно упругих столкновений между твердыми сферами. В 1960 году, возможно, при первом реалистичном моделировании материи, Гибсон и др. смоделировали радиационное повреждение твердой меди с помощью отталкивающего взаимодействия типа Борна – Майера наряду с силой сцепления на поверхности. В 1964 году Рахман опубликовал моделирование жидкого аргона с использованием потенциала Леннарда-Джонса ; расчеты свойств системы, таких как коэффициент самодиффузии, хорошо сравниваются с экспериментальными данными.
Метод МД, впервые использованный в теоретической физике, вскоре приобрел популярность в материаловедении, а с 1970-х годов также широко используется в биохимии и биофизике. МД часто используется для уточнения трехмерных структур белков и других макромолекул на основе экспериментальных ограничений рентгеновской кристаллографии или ЯМР-спектроскопии. В физике MD используется для изучения динамики явлений на атомном уровне, которые нельзя наблюдать напрямую, таких как рост тонких пленок и ионная субплантация, а также для изучения физических свойств нанотехнологических устройств, которые еще не созданы или не могут быть созданы.. В биофизике и структурной биологии этот метод часто применяется для изучения движений макромолекул, таких как белки и нуклеиновые кислоты, что может быть полезно для интерпретации результатов определенных биофизических экспериментов и для моделирования взаимодействий с другими молекулами, например, при стыковке лигандов. В принципе, MD может быть использовано для прогнозирования Неэмпирического из структуры белка путем имитации складывания из полипептидной цепи от случайной катушки.
Результаты моделирования МД могут быть проверены путем сравнения с экспериментами по измерению молекулярной динамики, популярным методом из которых является спектроскопия ЯМР. Прогнозы структуры, полученные на основе МД, могут быть проверены с помощью экспериментов в масштабе всего сообщества в Критической оценке предсказания структуры белка ( CASP ), хотя исторически этот метод имел ограниченный успех в этой области. Майкл Левитт, который разделил Нобелевскую премию частично за применение MD к белкам, писал в 1999 году, что участники CASP обычно не использовали этот метод из-за «. .. основного затруднения молекулярной механики, а именно минимизации энергии или молекулярной динамики в целом. приводит к модели, которая меньше похожа на экспериментальную структуру ". Улучшения в вычислительных ресурсах, позволяющих использовать все более и более длинные траектории МД, в сочетании с современными улучшениями качества параметров силового поля, привели к некоторым улучшениям как в предсказании структуры, так и в уточнении модели гомологии, без достижение практической полезности в этих областях; многие считают параметры силового поля ключевой областью для дальнейшего развития.
Сообщалось о моделировании МД для разработки фармакофоров и дизайна лекарств. Например, Пинто и соавт. реализовали моделирование MD комплексов Bcl-Xl для расчета средних положений критических аминокислот, участвующих в связывании лиганда. С другой стороны, Carlson et al. реализовано моделирование молекулярной динамики для идентификации соединений, которые дополняют рецептор, вызывая минимальное нарушение конформации и гибкости активного сайта. Снимки белка с постоянными интервалами времени во время моделирования накладывались, чтобы идентифицировать консервативные области связывания (консервативные по крайней мере в трех из одиннадцати рамок) для развития фармакофора. Spyrakis et al. полагался на рабочий процесс моделирования MD, отпечатков пальцев для лигандов и белков (FLAP) и линейного дискриминантного анализа для определения наилучших конформаций лиганд-белок, которые выступают в качестве фармакофорных шаблонов, на основе ретроспективного ROC-анализа полученных фармакофоров. В попытке улучшить моделирование открытия лекарств на основе структуры, учитывая потребность во многих смоделированных соединениях, Хатмал и др. Предложили комбинацию МД-моделирования и анализа межмолекулярных контактов лиганд-рецептор для выявления критических межмолекулярных контактов (связывающих взаимодействий). из дублирующих в единый лиганд-белковый комплекс. Затем критические контакты можно преобразовать в модели фармакофора, которые можно использовать для виртуального скрининга.
Ограничения метода связаны с используемыми наборами параметров и с основными силовыми полями молекулярной механики. Один прогон МД-моделирования оптимизирует потенциальную энергию, а не свободную энергию белка, что означает, что не учитываются все энтропийные вклады в термодинамическую стабильность структуры белка, включая конформационную энтропию полипептидной цепи (основной фактор, дестабилизирующий структуру белка.) и гидрофобные эффекты (основные движущие силы сворачивания белков). Другой важный фактор - внутримолекулярные водородные связи, которые явно не включены в современные силовые поля, но описываются как кулоновские взаимодействия точечных зарядов атомов. Это грубое приближение, поскольку водородные связи имеют частично квантово-механическую и химическую природу. Кроме того, электростатическое взаимодействие, как правило, рассчитаны с использованием диэлектрической постоянной в вакууме, хотя окружающий водный раствор имеет значительно более высокую диэлектрическую проницаемость. Использование макроскопической диэлектрической проницаемости на малых межатомных расстояниях сомнительно. Наконец, взаимодействия Ван-дер-Ваальса в МД обычно описываются потенциалами Леннарда-Джонса, основанными на теории Фрица Лондона, которая применима только в вакууме. Однако все типы сил Ван-дер-Ваальса в конечном итоге имеют электростатическое происхождение и, следовательно, зависят от диэлектрических свойств окружающей среды. Прямое измерение сил притяжения между различными материалами (как постоянная Гамакера ) показывает, что «взаимодействие между углеводородами через воду составляет около 10% от взаимодействия через вакуум». Зависимость сил Ван-дер-Ваальса от окружающей среды не учитывается в стандартном моделировании, но может быть учтена путем разработки поляризуемых силовых полей.
Дизайн моделирования молекулярной динамики должен учитывать доступную вычислительную мощность. Размер моделирования ( n = количество частиц), временной шаг и общая продолжительность должны быть выбраны так, чтобы расчет можно было завершить в разумный период времени. Однако моделирование должно быть достаточно продолжительным, чтобы соответствовать временным масштабам изучаемых природных процессов. Чтобы сделать статистически достоверные выводы из моделирования, моделируемый временной интервал должен соответствовать кинетике естественного процесса. В противном случае это аналогично выводам о том, как человек ходит, глядя менее чем на один шаг. В большинстве научных публикаций о динамике белков и ДНК используются данные моделирования, охватывающие от наносекунд (10 -9 с) до микросекунд (10 -6 с). Для получения этих симуляций требуется от нескольких дней до процессорных лет. Параллельные алгоритмы позволяют распределять нагрузку между процессорами; Примером может служить алгоритм пространственного или силового разложения.
Во время классического МД-моделирования наиболее интенсивной задачей для процессора является оценка потенциала как функции внутренних координат частиц. В рамках этой энергетической оценки самой дорогой является несвязанная или нековалентная часть. В нотации Big O общие модели молекулярной динамики масштабируются, если все парные электростатические и ван-дер-ваальсовы взаимодействия должны быть учтены явно. Эти вычислительные затраты могут быть уменьшены за счет использования методов электростатики, таких как суммирование Эвальда с сеткой частиц (), частица-частица-частица-сетка ( P3M ) или хорошие методы сферического отсечения ().
Другой фактор, который влияет на общее время процессора, необходимое для моделирования, - это размер временного шага интеграции. Это промежуток времени между оценками потенциала. Временной шаг должен быть выбран достаточно малым, чтобы избежать ошибок дискретизации (т. Е. Меньшим, чем период, связанный с самой быстрой частотой колебаний в системе). Типичные временные интервалы для классических MD составляют порядка 1 фемтосекунды (10 -15 с). Это значение может быть расширено с помощью таких алгоритмов, как алгоритм ограничения SHAKE, который фиксирует колебания самых быстрых атомов (например, водорода) на месте. Также были разработаны методы множественной шкалы времени, которые позволяют увеличивать время между обновлениями более медленных сил дальнего действия.
Для моделирования молекул в растворителе следует выбирать между явным и неявным растворителем. Явные частицы растворителя (такие как модели воды TIP3P, SPC / E и SPC-f ) должны быть дорогостоящими расчетами с помощью силового поля, в то время как неявные растворители используют подход среднего поля. Использование явного растворителя требует больших вычислительных ресурсов и требует включения в моделирование примерно в десять раз больше частиц. Но зернистость и вязкость явного растворителя важны для воспроизведения определенных свойств молекул растворенного вещества. Это особенно важно для воспроизведения химической кинетики.
Во всех видах моделирования молекулярной динамики размер окна моделирования должен быть достаточно большим, чтобы избежать артефактов граничных условий. Граничные условия часто обрабатываются путем выбора фиксированных значений на краях (что может вызвать артефакты) или использования периодических граничных условий, при которых одна сторона моделирования возвращается к противоположной стороне, имитируя объемную фазу (что также может вызывать артефакты).
 Схематическое изображение выборки поверхности потенциальной энергии системы с помощью молекулярной динамики (красный цвет) по сравнению с методами Монте-Карло (синий цвет).
Схематическое изображение выборки поверхности потенциальной энергии системы с помощью молекулярной динамики (красный цвет) по сравнению с методами Монте-Карло (синий цвет). В микроканоническом ансамбле система изолирована от изменений молей (N), объема (V) и энергии (E). Это соответствует адиабатическому процессу без теплообмена. Траектория микроканонической молекулярной динамики может рассматриваться как обмен потенциальной и кинетической энергией при сохранении полной энергии. Для системы из N частиц с координатами и скоростями следующая пара дифференциальных уравнений первого порядка может быть записана в обозначениях Ньютона как
Функция потенциальной энергии системы является функцией координат частицы. Его называют просто потенциалом в физике или силовым полем в химии. Первое уравнение происходит из законов движения Ньютона ; сила, действующая на каждую частицу в системе, может быть рассчитана как отрицательный градиент.
Для каждого временного шага положение и скорость каждой частицы могут быть интегрированы с помощью метода симплектического интегратора, такого как интегрирование Верле. Временная эволюция и называется траекторией. Учитывая начальные положения (например, из теоретических знаний) и скорости (например, рандомизированный гауссовский), мы можем вычислить все будущие (или прошлые) положения и скорости.
Одним из частых источников путаницы является значение температуры в МД. Обычно у нас есть опыт работы с макроскопическими температурами, в которых участвует огромное количество частиц. Но температура - это статистическая величина. Если имеется достаточно большое количество атомов, статистическую температуру можно оценить по мгновенной температуре, которую можно найти, приравняв кинетическую энергию системы к nk B T / 2, где n - количество степеней свободы системы.
Явление, связанное с температурой, возникает из-за небольшого количества атомов, которые используются в МД-моделировании. Например, рассмотрим моделирование роста пленки меди, начиная с подложки, содержащей 500 атомов, и с энергией осаждения 100 эВ. В реальном мире 100 эВ от осажденного атома быстро переносятся и распределяются между большим количеством атомов ( или более) без больших изменений температуры. Однако, когда имеется только 500 атомов, подложка почти сразу испаряется в результате осаждения. Нечто подобное происходит в биофизическом моделировании. Температура системы в NVE естественно повышается, когда макромолекулы, такие как белки, подвергаются экзотермическим конформационным изменениям и связыванию.
В каноническом ансамбле сохраняются количество вещества (N), объем (V) и температура (T). Его также иногда называют молекулярной динамикой при постоянной температуре (CTMD). В NVT энергия эндотермических и экзотермических процессов обменивается с термостатом.
Доступны различные алгоритмы термостата для добавления и удаления энергии из границ моделирования МД более или менее реалистичным способом, приближая канонический ансамбль. Популярные методы для контроля температуры включают скорость масштабирование, в термостат Nose-Hoover, Nose-Hoover цепи, термостат Берендсен, то термостат Андерсен и динамика Ланжевенна. Термостат Берендсена может вызвать эффект летающего кубика льда, который приводит к нефизическим перемещениям и поворотам моделируемой системы.
Используя эти алгоритмы, нетривиально получить каноническое ансамблевое распределение конформаций и скоростей. Как это зависит от размера системы, выбора термостата, параметров термостата, временного шага и интегратора - тема многих статей в данной области.
В изотермически-изобарическом ансамбле сохраняются количество вещества (N), давление (P) и температура (T). Помимо термостата нужен баростат. Это наиболее близко соответствует лабораторным условиям с колбой, открытой для температуры и давления окружающей среды.
При моделировании биологических мембран изотропный контроль давления не подходит. Для липидных бислоев регулирование давления происходит при постоянной площади мембраны (NPAT) или постоянном поверхностном натяжении «гамма» (NPγT).
Метод обмена репликами представляет собой обобщенный ансамбль. Первоначально он был создан для изучения медленной динамики неупорядоченных спиновых систем. Его также называют параллельным отпуском. Формулировка MD с заменой реплик (REMD) пытается преодолеть проблему множественных минимумов, меняя температуру невзаимодействующих реплик системы, работающей при нескольких температурах.
Моделирование молекулярной динамики требует определения потенциальной функции или описания условий, с помощью которых частицы в моделировании будут взаимодействовать. В химии и биологии это обычно называют силовым полем, а в физике материалов - межатомным потенциалом. Потенциалы могут быть определены на многих уровнях физической точности; те, которые наиболее часто используются в химии, основаны на молекулярной механике и воплощают классическую механическую трактовку взаимодействий между частицами, которые могут воспроизводить структурные и конформационные изменения, но обычно не могут воспроизводить химические реакции.
Сведение от полностью квантового описания к классическому потенциалу влечет за собой два основных приближения. Первое - это приближение Борна – Оппенгеймера, которое утверждает, что динамика электронов настолько быстра, что можно считать, что они мгновенно реагируют на движение своих ядер. Как следствие, их можно рассматривать отдельно. Второй трактует ядра, которые намного тяжелее электронов, как точечные частицы, которые следуют классической ньютоновской динамике. В классической молекулярной динамике влияние электронов аппроксимируется одной поверхностью потенциальной энергии, обычно представляющей основное состояние.
Когда требуются более тонкие уровни детализации, используются потенциалы, основанные на квантовой механике ; некоторые методы пытаются создать гибридные классические / квантовые потенциалы, в которых основная часть системы рассматривается классически, а небольшая область рассматривается как квантовая система, обычно подвергающаяся химическому преобразованию.
Эмпирические потенциалы, используемые в химии, часто называют силовыми полями, а те, которые используются в физике материалов, - межатомными потенциалами.
Большинство силовых полей в химии являются эмпирическими и состоят из суммы связанных сил, связанных с химическими связями, валентными углами и двугранными связями, и несвязанных сил, связанных с силами Ван-дер-Ваальса и электростатическим зарядом. Эмпирические потенциалы представляют квантово-механические эффекты ограниченным образом через специальные функциональные приближения. Эти потенциалы содержат свободные параметры, такие как атомный заряд, параметры Ван-дер-Ваальса, отражающие оценки атомного радиуса и равновесной длины связи, угла и двугранности; они получены путем сопоставления с подробными электронными расчетами (квантово-химическое моделирование) или экспериментальными физическими свойствами, такими как упругие постоянные, параметры решетки и спектроскопические измерения.
Из-за нелокальной природы несвязанных взаимодействий они включают, по крайней мере, слабые взаимодействия между всеми частицами в системе. Его расчет обычно является узким местом в скорости моделирования МД. Чтобы снизить вычислительные затраты, силовые поля используют численные приближения, такие как смещенные радиусы отсечки, алгоритмы поля реакции, суммирование Эвальда сетки частиц или новую сетку частица-частица-частица-сетка ( P3M ).
Химические силовые поля обычно используют заранее заданные схемы связывания (исключение составляет ab initio динамика) и, таким образом, не могут в явном виде моделировать процесс разрыва химической связи и реакции. С другой стороны, многие из потенциалов, используемых в физике, например, основанные на формализме порядка связи, могут описывать несколько различных координаций системы и разрыв связей. Примеры таких потенциалов включают потенциал Бреннера для углеводородов и его дальнейшие разработки для систем C-Si-H и COH. ReaxFF потенциал можно считать полностью реактивным гибрид между потенциалами порядка связи и полой химия силы.
Потенциальные функции, представляющие несвязанную энергию, формулируются как сумма взаимодействий между частицами системы. Самый простой выбор, используемый во многих популярных силовых полях, - это «парный потенциал», в котором полную потенциальную энергию можно вычислить из суммы вкладов энергии между парами атомов. Поэтому эти силовые поля также называют «аддитивными силовыми полями». Примером такого парного потенциала является несвязанный потенциал Леннарда – Джонса (также называемый потенциалом 6–12), используемый для расчета сил Ван-дер-Ваальса.
Другой пример - борновская (ионная) модель ионной решетки. Первый член в следующем уравнении - это закон Кулона для пары ионов, второй член - это короткодействующее отталкивание, объясняемое принципом исключения Паули, а последний член - член дисперсионного взаимодействия. Обычно моделирование включает только диполярный член, хотя иногда также включается квадрупольный член. При n l = 6 этот потенциал также называют потенциалом Кулона – Букингема.
В потенциалах многих тел потенциальная энергия включает эффекты трех или более частиц, взаимодействующих друг с другом. При моделировании с парными потенциалами глобальные взаимодействия в системе также существуют, но они происходят только через парные члены. В многочастичных потенциалах потенциальная энергия не может быть найдена суммой по парам атомов, поскольку эти взаимодействия вычисляются явно как комбинация членов более высокого порядка. Со статистической точки зрения, зависимость между переменными, как правило, не может быть выражена с использованием только попарных произведений степеней свободы. Например, потенциал Терсоффа, который первоначально использовался для моделирования углерода, кремния и германия и с тех пор используется для широкого круга других материалов, включает в себя сумму по группам из трех атомов, причем углы между атомами равны важный фактор в потенциале. Другими примерами являются потенциалы метода встроенного атома (EAM), EDIP и аппроксимации второго момента сильной связи (TBSMA), где плотность состояний электронов в области атома вычисляется из суммы вкладов от окружающих атомов., и тогда вклад потенциальной энергии является функцией этой суммы.
Полуэмпирические потенциалы используют матричное представление из квантовой механики. Однако значения матричных элементов находятся с помощью эмпирических формул, оценивающих степень перекрытия конкретных атомных орбиталей. Затем матрица диагонализируется для определения степени занятости различных атомных орбиталей, и эмпирические формулы снова используются для определения энергетических вкладов орбиталей.
Существует множество полуэмпирических потенциалов, называемых потенциалами сильной связи, которые варьируются в зависимости от моделируемых атомов.
Большинство классических силовых полей неявно включают эффект поляризуемости, например, путем увеличения частичных зарядов, полученных из квантово-химических расчетов. Эти частичные заряды стационарны по отношению к массе атома. Но моделирование молекулярной динамики может явно моделировать поляризуемость с введением индуцированных диполей с помощью различных методов, таких как частицы Друде или флуктуирующие заряды. Это позволяет динамически перераспределять заряд между атомами, реагируя на локальное химическое окружение.
В течение многих лет моделирование поляризуемых МД рекламировалось как следующее поколение. Для однородных жидкостей, таких как вода, повышенная точность была достигнута за счет включения поляризуемости. Некоторые многообещающие результаты были достигнуты и в отношении белков. Однако все еще не ясно, как лучше всего аппроксимировать поляризуемость при моделировании.
В классической молекулярной динамике одна поверхность потенциальной энергии (обычно основное состояние) представлена в силовом поле. Это следствие приближения Борна – Оппенгеймера. В возбужденных состояниях, химических реакциях или когда требуется более точное представление, электронное поведение может быть получено из первых принципов с помощью квантово-механического метода, такого как теория функционала плотности. Это называется Ab Initio Molecular Dynamics (AIMD). Из-за стоимости обработки электронных степеней свободы вычислительные затраты на это моделирование намного выше, чем при классической молекулярной динамике. Это означает, что AIMD ограничивается меньшими системами и более коротким временем.
Ab initio квантово-механические и химические методы могут использоваться для вычисления потенциальной энергии системы на лету, что необходимо для конформаций в траектории. Этот расчет обычно производится в непосредственной близости от координаты реакции. Хотя могут использоваться различные приближения, они основаны на теоретических соображениях, а не на эмпирической подгонке. Расчеты ab initio дают огромное количество информации, недоступной с помощью эмпирических методов, например, плотности электронных состояний или других электронных свойств. Существенным преимуществом использованияметодов ab initio является возможность изучать реакции, которые включают разрыв или образование ковалентных связей, которые соответствуют множественным электронным состояниям. Более того, неэмпирические методы также позволяют восстанавливать эффекты, выходящие за рамки приближения Борна – Оппенгеймера, используя такие подходы, как смешанная квантово-классическая динамика.
Квантово-механические методы очень эффективны. Однако они дорогостоящие в вычислительном отношении, в то время как методы MM (классической или молекулярной механики) быстры, но имеют несколько ограничений (требуют обширной параметризации; полученные оценки энергии не очень точны; не могут использоваться для моделирования реакций, в которых ковалентные связи разрываются / образуются. ; и ограничены в своих возможностях по предоставлению точных сведений о химической среде). Появился новый класс методов, сочетающий в себе достоинства расчетов QM (точность) и MM (скорость). Эти методы называются смешанными или гибридными методами квантовой механики и молекулярной механики (гибридный QM / MM).
Важнейшим преимуществом гибридного метода QM / MM является скорость. Стоимость выполнения классической молекулярной динамики (ММ) в самом простом случае составляет O (n 2), где n - количество атомов в системе. Это в основном связано с термином электростатического взаимодействия (каждая частица взаимодействует с каждой другой частицей). Однако использование радиуса отсечки, периодических обновлений парных списков и недавних вариаций метода Эвальда (PME) с частичной сеткой уменьшило это значение до O (n) до O (n 2). Другими словами, если смоделировать систему с вдвое большим количеством атомов, то потребуется от двух до четырех раз больше вычислительной мощности. С другой стороны, простейшие расчеты ab initio обычно имеют масштаб O (n 3) или хуже (ограниченные вычисления Хартри-Фока предлагались с масштабом ~ O (n 2,7)). Чтобы преодолеть этот предел, небольшая часть системы обрабатывается квантово-механически (обычно активный центр фермента), а оставшаяся система обрабатывается классическим способом.
В более сложных реализациях существуют методы QM / MM для лечения как легких ядер, чувствительных к квантовым эффектам (например, водородов), так и электронных состояний. Это позволяет генерировать волновые функции водорода (аналогичные электронным волновым функциям). Эта методология была полезна при исследовании таких явлений, как туннелирование водорода. Одним из примеров, когда методы QM / MM предоставили новые открытия, является расчет переноса гидрида в ферменте алкогольдегидрогеназе печени. В этом случае для водорода важно квантовое туннелирование, так как оно определяет скорость реакции.
На другом конце шкалы детализации находятся крупнозернистые и решетчатые модели. Вместо явного представления каждого атома системы для представления групп атомов используются «псевдоатомы». МД-моделирование на очень больших системах может потребовать таких больших компьютерных ресурсов, что их нелегко изучить традиционными полностью атомными методами. Точно так же моделирование процессов в длительных масштабах времени (более 1 микросекунды) непомерно дорого, потому что для этого требуется очень много временных шагов. В этих случаях иногда можно решить проблему, используя сокращенные представления, которые также называются крупнозернистыми моделями.
Примерами крупнозернистых (CG) методов являются разрывная молекулярная динамика (CG-DMD) и Go-модели. Иногда выполняется крупнозернистая обработка псевдоатомов большего размера. Такие приближения объединенного атома использовались в МД-моделировании биологических мембран. Реализация такого подхода в системах, где электрические свойства представляют интерес, может быть сложной задачей из-за сложности использования правильного распределения заряда на псевдоатомах. Алифатические хвосты липидов представлены несколькими псевдоатомами, объединяющими от 2 до 4 метиленовых групп в каждый псевдоатом.
Параметризация этих очень грубых моделей должна выполняться эмпирически, согласовывая поведение модели с соответствующими экспериментальными данными или полностью атомным моделированием. В идеале эти параметры должны неявно учитывать как энтальпийный, так и энтропийный вклады в свободную энергию. Когда грубая обработка выполняется на более высоких уровнях, точность динамического описания может быть менее надежной. Но очень крупнозернистые модели успешно использовались для изучения широкого круга вопросов структурной биологии, организации жидких кристаллов и полимерных стекол.
Примеры применения крупнозернистого зерна:
Простейшая форма крупнозернистой структуры - объединенный атом (иногда называемый протяженным атомом) - использовалась в большинстве ранних моделей МД белков, липидов и нуклеиновых кислот. Например, вместо того, чтобы рассматривать все четыре атома метильной группы CH 3 явно (или все три атома метиленовой группы CH 2), один представляет всю группу с одним псевдоатомом. Конечно, он должен быть правильно параметризован, чтобы его ван-дер-ваальсовы взаимодействия с другими группами имели правильную зависимость от расстояния. Аналогичные соображения применимы к связям, углам и кручениям, в которых участвует псевдоатом. В таком виде представления объединенного атома обычно исключаются все явные атомы водорода, кроме тех, которые обладают способностью участвовать в водородных связях ( полярные водороды). Примером этого является силовое поле CHARMM 19.
Полярные водороды обычно сохраняются в модели, потому что надлежащая обработка водородных связей требует достаточно точного описания направленности и электростатических взаимодействий между донорной и акцепторной группами. Например, гидроксильная группа может быть как донором водородной связи, так и акцептором водородной связи, и было бы невозможно обработать это одним псевдоатомом ОН. Около половины атомов в белке или нуклеиновой кислоте являются неполярными атомами водорода, поэтому использование объединенных атомов может обеспечить значительную экономию компьютерного времени.
Во многих симуляциях системы растворенное вещество-растворитель основное внимание уделяется поведению растворенного вещества без особого интереса к поведению растворителя, особенно в тех молекулах растворителя, которые находятся в областях, далеких от молекулы растворенного вещества. Растворители могут влиять на динамическое поведение растворенных веществ посредством случайных столкновений и создания сопротивления трения движению растворенных веществ через растворитель. Использование непрямоугольных периодических граничных условий, стохастических границ и оболочек растворителя может помочь уменьшить количество требуемых молекул растворителя и позволить большую часть вычислительного времени тратить на моделирование растворенного вещества. Также возможно включить эффекты растворителя без необходимости явного присутствия молекул растворителя. Одним из примеров этого подхода является использование потенциальной средней силы (PMF), которая описывает, как изменяется свободная энергия при изменении конкретной координаты. Изменение свободной энергии, описываемое PMF, содержит усредненные эффекты растворителя.
Взаимодействие на больших расстояниях - это взаимодействие, при котором пространственное взаимодействие спадает не быстрее, чем где - размерность системы. Примеры включают заряд-зарядовые взаимодействия между ионами и диполь-дипольные взаимодействия между молекулами. Моделирование этих сил представляет собой довольно сложную задачу, поскольку они значительны на расстоянии, которое может превышать половину длины ящика при моделировании многих тысяч частиц. Хотя одним из решений было бы значительно увеличить размер блока, этот метод грубой силы далеко не идеален, поскольку моделирование стало бы очень дорогостоящим в вычислительном отношении. Сферическое усечение потенциала также исключено, поскольку может наблюдаться нереалистичное поведение, когда расстояние близко к расстоянию отсечения.
Моделирование управляемой молекулярной динамики (SMD) или моделирование силового зонда применяет силы к белку, чтобы манипулировать его структурой, перемещая его по желаемым степеням свободы. Эти эксперименты могут быть использованы для выявления структурных изменений белка на атомном уровне. SMD часто используется для моделирования таких событий, как механическое разворачивание или растяжение.
Существует два типичных протокола SMD: один, в котором скорость вытягивания поддерживается постоянной, и второй, в котором приложенная сила постоянна. Обычно часть исследуемой системы (например, атом в белке) сдерживается гармоническим потенциалом. Затем силы применяются к конкретным атомам либо с постоянной скоростью, либо с постоянной силой. Зонтичная выборка используется для перемещения системы по желаемой координате реакции путем изменения, например, сил, расстояний и углов, которыми манипулируют при моделировании. Благодаря зонтичной выборке все конфигурации системы - как высокоэнергетические, так и низкоэнергетические - отбираются надлежащим образом. Затем изменение свободной энергии каждой конфигурации можно рассчитать как потенциал средней силы. Популярным методом вычисления PMF является метод анализа взвешенной гистограммы (WHAM), который анализирует серию имитаций зонтичной выборки.
Многие важные приложения SMD находятся в области открытия лекарств и биомолекулярных наук. Например, SMD использовался для исследования стабильности протофибрилл Альцгеймера, для изучения взаимодействия белкового лиганда в циклин-зависимой киназе 5 и даже для демонстрации влияния электрического поля на тромбин (белок) и аптамерный (нуклеотидный) комплекс среди многих других интересных исследований..
 Молекулярно-динамическое моделирование синтетического молекулярного двигателя, состоящего из трех молекул в нанопоре (внешний диаметр 6,7 нм) при 250 К.
Молекулярно-динамическое моделирование синтетического молекулярного двигателя, состоящего из трех молекул в нанопоре (внешний диаметр 6,7 нм) при 250 К. Молекулярная динамика используется во многих областях науки.
Следующие биофизические примеры иллюстрируют заметные усилия по моделированию систем очень большого размера (полный вирус) или очень длительного времени моделирования (до 1,112 миллисекунд):
Еще одно важное применение метода МД извлекает выгоду из его способности к трехмерной характеристике и анализу эволюции микроструктуры в атомном масштабе.












![U (r) = 4 \ varepsilon \ left [\ left (\ frac {\ sigma} {r} \ right) ^ {12} - \ left (\ frac {\ sigma} {r} \ right) ^ {6} \Правильно]](https://wikimedia.org/api/rest_v1/media/math/render/svg/374024e23ac5eb77e91b68ad9ba86ad3bbf5f113)


