
Войти
| Гемофагоцитарный лимфогистиоцитоз | |
|---|---|
| Другие названия | HLH |
 | |
| Микрофотография с эритроцитами внутри макрофагов. Окраска HE. | |
| Специальность | Гематология |
Гемофагоцитарный лимфогистиоцитоз (HLH ), также известный как гемофагоцитарный лимфогистиоцитоз (британское написание ), и гемофагоцитарный или гемофагоцитарный синдром, это необычное гематологическое заболевание, которое чаще встречается у детей, чем у взрослых. Это опасное для жизни заболевание тяжелого гипервоспаления, вызванное неконтролируемой пролиферацией активированных лимфоцитов и макрофагов, характеризующееся пролиферацией морфологически доброкачественных лимфоцитов и макрофагов, которые секретируют большое количество количества воспалительных цитокинов. Он классифицируется как один из синдромов цитокинового шторма. Различают наследственные и ненаследственные (приобретенные) причины гемофагоцитарного лимфогистиоцитоза (ГЛГ).
Начало ГЛГ происходит в возрасте до одного года примерно в 70 процентах случаев. Следует заподозрить семейный ГЛГ, если у братьев и сестер диагностирован ГЛГ или если симптомы повторяются после прекращения терапии. У каждого полноправного брата или сестры ребенка с семейным HLH есть 25% -ный шанс развития болезни, 50% -ный шанс быть носителем дефектного гена (что очень редко связано с каким-либо риском заболевания) и 25%. –Процентная вероятность того, что они не затронуты и не будут нести дефект гена.
Пациентам с ГЛГ, особенно без лечения, может потребоваться интенсивная терапия. Следовательно, HLH следует включать в дифференциальную диагностику пациентов отделения интенсивной терапии с цитопенией и гиперферритинемией. Пациенты на ранних стадиях ГЛГ часто госпитализируются в отделения внутренней медицины.
ВГВ клинически проявляется лихорадкой, увеличением печени и селезенки, увеличенные лимфатические узлы, изменение цвета кожи и глаз на желтый цвет и сыпь. Лабораторные данные могут включать повышенный уровень триглицеридов, низкий уровень фибриногена, трансаминит и повышенный уровень ферритина (среди прочего).
Первичный HLH вызван потерей функции (т. Е. инактивация) мутаций в генах, кодирующих белки цитотоксические Т-клетки и NK-клетки, используемые для уничтожения клеток-мишеней, например, инфицированных патогенами, такими как вирус Эпштейна-Барра (EBV) или вирус денге. Эти мутации включают мутации в следующих генах: UNC13D, STX11, RAB27A, STXBP2, LYST, PRF1 1, SH2D1A, BIRC4, ITK, CD27 и MAGT1.
вторичный HLH (sHLH) связан с злокачественными и незлокачественными заболеваниями, которые также ослабляют способность иммунной системы атаковать EBV-инфицированные клетки и, как считается, им способствуют. Злокачественные заболевания, связанные с вторичным HLH, включают Т-клеточную лимфому, В-клеточную лимфому, острый лимфоцитарный лейкоз, острый миелоидный лейкоз и миелодиспластический синдром. Незлокачественные заболевания, связанные с вторичным HLH, включают: аутоиммунные нарушения, такие как ювенильный идиопатический артрит, ювенильный болезнь Кавасаки, системная красная волчанка, ювенильное начало. и взрослые формы болезни Стилла и ревматоидный артрит ; иммунодефицитные расстройства, такие как тяжелый комбинированный иммунодефицит, Синдром ДиДжорджи, синдром Вискотта – Олдрича, атаксия – телеангиэктазия и врожденный дискератоз ); и инфекции, вызванные EBV, цитомегаловирусом, ВИЧ / СПИДом, бактериями, простейшими, грибами и, возможно, SARS-CoV-2. Вторичный HLH также может быть результатом ятрогенных причин, таких как трансплантация костного мозга или других органов; химиотерапия; или терапия иммунодепрессантами;
Около 33% всех случаев ГЛГ, ~ 75% случаев ГЛГ в Азии и почти 100% случаев ГЛГ, вызванных мутациями в SH2D1A (см. Х-сцепленное лимфопролиферативное заболевание тип 1 ) связаны с ВЭБ-инфекцией и считаются вызванными ею. Эти случаи HLH классифицируются как принадлежащие к классу лимфопролиферативных заболеваний, связанных с вирусом Эпштейна – Барра и обозначаются EBV + HLH.
Основные причины, наследуемые или приобретенные, приводят к неконтролируемому иммунному ответу при воздействии триггеров. Нарушение цитотоксичности NK-клеток является отличительной чертой HLH. Все генетические дефекты семейного HLH связаны с гранульной -зависимой цитотоксичностью. Эта неспособность удалить инфицированные и антигенпрезентирующие клетки и прекратить иммунный ответ приводит к неконтролируемой пролиферации и активации иммунной системы с высвобождением избыточных цитокинов. Затем эти клетки проникают в органы, высвобождая больше цитокинов, что дает клиническую картину. Лихорадка вызывается IL-1, IL-6 и TNF-alpha ; цитопения возникает из-за подавляющего действия TNF-альфа и TNF-гамма на гемопоэз. TNF-альфа и TNF-гамма также могут приводить к ингибированию липопротеин липазы или стимулировать синтез триглицеридов. Активированные макрофаги секретируют ферритин и активатор плазминогена, что приводит к гиперфибринолизу.
Пять генетических подтипов (FHL1, FHL2, FHL3, FHL4 и FHL5) описаны с оценочной общей распространенностью один на 50 000 и равным гендерным распределением. Молекулярно-генетическое тестирование четырех причинных генов, PRF1 (FHL2), UNC13D (FHL3), STX11 (FHL4) и STXBP2 (FHL5), доступно на клинической основе. Симптомы FHL обычно проявляются в течение первых нескольких месяцев жизни и могут даже развиться внутриутробно. Однако в некоторых случаях наблюдаются симптоматические проявления в детстве и даже в молодом зрелом возрасте.
Каждый из пяти подтипов FHL связан с определенным геном:
Почти половина случаев семейного гемофагоцитарного лимфогистиоцитоза 2 типа обусловлена биаллельным Мутации PRF1.
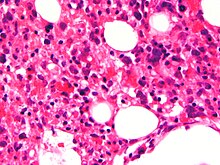 Изображение костного мозга с помощью светового микроскопа, показывающее стромальные макрофаги, содержащие многочисленные эритроциты в своей цитоплазме
Изображение костного мозга с помощью светового микроскопа, показывающее стромальные макрофаги, содержащие многочисленные эритроциты в своей цитоплазме Анализ крови обычно показывает уменьшение количества клеток крови, включая уменьшение количества циркулирующих эритроцитов, лейкоцитов и тромбоцитов. Костный мозг может показать гемофагоцитоз. Функциональные пробы печени обычно повышены. Низкий уровень o f белок альбумин в крови встречается часто.
Сывороточный C-реактивный белок, скорость оседания эритроцитов и ферритин уровень заметно приподнят. У детей ферритин выше 10000 очень чувствителен и специфичен для диагностики ГЛГ, однако диагностическая ценность ферритина меньше для взрослых пациентов с ГЛГ.
Уровень фибриногена в сыворотке обычно составляет низкий и уровень D-димера повышен.
сфингомиелиназа повышена.
Биопсия костного мозга показывает гистиоцитоз.
Первичный HLH, также известный как Семейный гемофагоцитарный лимфогистиоцитоз (FHL) или семейный эритрофагоцитарный лимфогистиоцитоз - это гетерогенное аутосомно-рецессивное заболевание, которое чаще встречается при родительском кровном родстве.
Вторичная гемофагоцитимоцитозофизофизофизическая активация (такая сильная иммунофагоцитозофизофизитическая активация) возникает после сильной иммунофагоцитозофизофизической активации (такая приобретенная) как то, что может происходить при системной инфекции, иммунодефиците или злокачественном новообразовании.
Обе формы характеризуются подавляющей активацией нормальных Т-лимфоцитов и макрофагов, что неизменно приводит к клиническим и гематологическим изменениям и смерти в отсутствие лечения.
Описан подтип первичного HLH, при котором воспаление ограничивается центральной нервной системой.
Текущая диагностика (2008 г.) Критерии HLH:
1. Молекулярный диагноз, соответствующий HLH. Они включают идентификацию патологических мутаций PRF1, UNC13D или STX11.
ИЛИ
2. Выполнение пяти из восьми критериев ниже:
Кроме того, в случае семейного HLH нет признаков злокачественности должно быть очевидным.
Не все пять критериев из восьми необходимы для диагностики ГЛГ у взрослых, и высокий индекс s Успехи необходимы для постановки диагноза, так как задержка приводит к увеличению смертности. Диагностические критерии были разработаны в педиатрической популяции и не были валидированы для взрослых пациентов с HLH. Попытки улучшить диагностику HLH включали использование HScore, который можно использовать для оценки индивидуального риска HLH. Было обнаружено, что у взрослых растворимый ИЛ-2 является очень чувствительным маркером ГЛГ, демонстрируя 100% чувствительность для исключения ГЛГ ниже порогового значения 2400 Ед / мл и оптимального порогового значения для исключения при 2515 Ед / мл (чувствительность 100 %; специфичность 72,5%) со специфичностью 93% при>10 000 Ед / мл.
Дифференциальный диагноз ГЛГ включает вторичный ГЛГ и активацию макрофагов синдром или другие первичные иммунодефициты, которые проявляются гемофагоцитарным лимфогистиоцитозом, такие как Х-сцепленное лимфопролиферативное заболевание.
Другие состояния, которые можно спутать с этим состоянием, включают аутоиммунный лимфопролиферативный синдром. Как синдром интенсивного воспаления его необходимо дифференцировать от сепсиса, что может быть чрезвычайно сложно.
Диагноз приобретенного или вторичного ГЛГ обычно ставится в связи с заражением вирусами., бактерии, грибки или паразиты, или в связи с лимфомой, аутоиммунным заболеванием или метаболическим заболеванием. Приобретенный ГЛГ может иметь пониженную, нормальную или повышенную активность NK-клеток.
Основным дифференциальным диагнозом ГЛГ является синдром Гричелли ( тип 2). Это редкое аутосомно-рецессивное заболевание, характеризующееся частичным альбинизмом, гепатоспленомегалией, панцитопенией, гепатитом, иммунологическими аномалиями и лимфогистиоцитозом. Большинство случаев было диагностировано в возрасте от 4 месяцев до 7 лет, средний возраст - около 17 месяцев.
Различают три типа синдрома Гричелли: тип 1 имеет неврологические симптомы и мутации в MYO5A. Прогноз зависит от тяжести неврологических проявлений. Тип 2 имеет мутации в RAB27A и гемофагоцитарный синдром с аномальной активацией Т-клеток и макрофагов. При отсутствии лечения у этого типа есть тяжелый прогноз. Тип 3 имеет мутации в меланофилине и характеризуется частичным альбинизмом. Этот тип не представляет угрозы для пострадавших.
Во вторичных случаях показано лечение причины, где это возможно. Кроме того, обычно требуется лечение самого HLH.
Хотя оптимальное лечение ГЛГ все еще обсуждается, текущие схемы лечения обычно включают высокие дозы кортикостероидов, этопозид и циклоспорин. Также используется внутривенный иммуноглобулин. Метотрексат и винкристин также использовались. Другие лекарства включают.
20 ноября 2018 г. FDA одобрило моноклональное антитело против IFN-гамма эмапалумаб (собственное название Gamifant) для лечения первичного HLH у детей и взрослых.
Прогноз безопасный, общая летальность составляет 50%. К неблагоприятным прогностическим факторам относились HLH, связанный со злокачественными новообразованиями, при этом половина пациентов умирала через 1,4 месяца по сравнению с 22,8 месяцами для пациентов с HLH, не связанных с опухолью.
Вторичный HLH у некоторых людей может быть ограничен самостоятельно, поскольку пациенты могут полностью выздороветь после получения только поддерживающего лечения (т. е. только внутривенного введения иммуноглобулина ). Однако длительная ремиссия без использования цитотоксической и иммуносупрессивной терапии маловероятна у большинства взрослых с ГЛГ и у пациентов с поражением центральной нервной системы (головного и / или спинного мозга).
Первое сообщение о случае HLH было опубликовано в 1952 году.
В недавно проведенном систематическом обзоре сообщалось, что объединенная доля - это лихорадка 97,2%, гепатомегалия 70,2%, спленомегалия 78,4%, тромбоцитопения 90,1%, анемия 76,0% и ферритин сыворотки ≥500 мкг / л 97,1%. Летальность среди пациентов с гемофагоцитарным лимфогистиоцитозом денге составляет 14,6%.
| Классификация | D |
|---|---|
| Внешние ресурсы |
.