
Войти
| Протеопатия | |
|---|---|
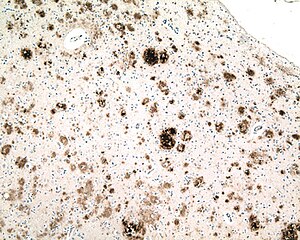 | |
| Микрофотография части коры головного мозга от человека с Болезнь Альцгеймера, иммуноокрашенная антителом к бета-амилоиду (коричневый), фрагмент белка, который накапливается в сенильных бляшках и церебральная амилоидная ангиопатия. Объектив микроскопа 10X. |
В медицине, протеопатии (; от протео- [предпочтительный белок]; -патия [суточное заболевание] ; протеопатии pl.; протеопатические прил.) Относится к классу заболеваний, при которых определенные белки становятся структурно аномальными и тем самым нарушают функция клеток, тканей и органов тела. Часто белки не могут свернуться до своей нормальной конфигурации ; в этом неправильно свернутом состоянии белки могут стать каким-то образом токсичными (токсичное усиление функции ) или они могут потерять свою нормальную функцию. Протеопатии (также известные как протеинопатии, белковые конформационные нарушения или болезни неправильного сворачивания белков ) включают такие заболевания, как болезнь Крейтцфельда-Якоба и другие прионные заболевания, болезнь Альцгеймера, болезнь Паркинсона, амилоидоз, множественная системная атрофия и широкий спектр других расстройств. Термин протеопатия был впервые предложен в 2000 году Лэри Уокером и Гарри Левином.
Концепция протеопатии может проследить свое происхождение до середины XIX века, когда в 1854 году Рудольф Вирхов ввел термин амилоид («крахмалоподобный») для описания вещества в мозговых телах amylacea, которое проявляет химическую реакцию, напоминающую реакцию целлюлозы. В 1859 году Фридрейх и Кекуле продемонстрировали, что «амилоид» не состоит из целлюлозы, а на самом деле богат белком. Последующие исследования показали, что многие различные белки могут образовывать амилоид, и что все амилоиды проявляют двойное лучепреломление в кросс- поляризованном свете после окрашивания красителем конго красным, поскольку а также фибриллярная ультраструктура при просмотре в электронном микроскопе. Однако некоторые белковые образования лишены двойного лучепреломления и содержат мало или совсем не содержат классических амилоидных фибрилл, например, диффузные отложения бета-амилоидного (Aβ) белка в мозге людей с болезнью Альцгеймера. Кроме того, появились доказательства того, что небольшие нефибриллярные белковые агрегаты, известные как олигомеры, токсичны для клеток пораженного органа, и что амилоидогенные белки в их фибриллярной форме могут быть относительно доброкачественными.
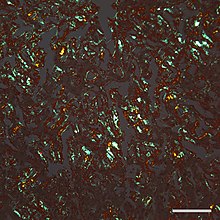 Микрофотография амилоид в срезе печени, который был окрашен красителем Конго красным и просматривался с помощью перекрестных поляризационных фильтров, давая типичное оранжево-зеленоватое двулучепреломление. Объектив микроскопа 20Х; масштабная линейка составляет 100 микрон (0,1 мм).
Микрофотография амилоид в срезе печени, который был окрашен красителем Конго красным и просматривался с помощью перекрестных поляризационных фильтров, давая типичное оранжево-зеленоватое двулучепреломление. Объектив микроскопа 20Х; масштабная линейка составляет 100 микрон (0,1 мм). В большинстве, если не во всех протеопатиях, изменение 3-мерного фолдинга (конформации) увеличивает тенденцию конкретного белка связываться с самим собой. В этой агрегированной форме белок устойчив к клиренсу и может нарушать нормальную способность пораженных органов. В некоторых случаях неправильная укладка белка приводит к потере его обычной функции. Например, кистозный фиброз вызывается дефектным белком-регулятором трансмембранной проводимости кистозного фиброза (CFTR), а при боковом амиотрофическом склерозе / лобно-височной долевой дегенерации (FTLD) - некоторыми ген-регулирующими белками. неправильно агрегируются в цитоплазме и, таким образом, не могут выполнять свои обычные задачи в ядре. Поскольку белки имеют общую структурную особенность, известную как основа полипептида, все белки могут при некоторых обстоятельствах неправильно складываться. Однако только относительно небольшое количество белков связано с протеопатическими нарушениями, возможно, из-за структурных идиосинкразий уязвимых белков. Например, белки, которые обычно развернуты или относительно нестабильны как мономеры (то есть как отдельные несвязанные белковые молекулы), с большей вероятностью будут неправильно свернуты в аномальную конформацию. Почти во всех случаях молекулярная конфигурация, вызывающая заболевание, включает увеличение вторичной структуры бета-листа белка. Было показано, что аномальные белки при некоторых протеопатиях складываются в несколько трехмерных форм; В этих вариантах белковые структуры определяются различными патогенными, биохимическими и конформационными свойствами. Они были наиболее тщательно изучены в отношении прионной болезни и называются штаммами белков .
 . Микрофотография иммуноокрашенного α-синуклеина (коричневый) в Тельца Леви (большие скопления) и нейриты Леви (нитевидные структуры) в коре головного мозга пациента с болезнью с тельцами Леви, синуклеинопатией. Объектив микроскопа 40X.
. Микрофотография иммуноокрашенного α-синуклеина (коричневый) в Тельца Леви (большие скопления) и нейриты Леви (нитевидные структуры) в коре головного мозга пациента с болезнью с тельцами Леви, синуклеинопатией. Объектив микроскопа 40X. Вероятность развития протеопатии увеличивается из-за определенных факторов риска, которые способствуют самосборке белка. К ним относятся дестабилизирующие изменения в первичной аминокислотной последовательности белка, посттрансляционные модификации (такие как гиперфосфорилирование ), изменения температуры или pH., увеличение выработки белка или уменьшение его клиренса. Пожилой возраст является серьезным фактором риска, как и черепно-мозговая травма. В стареющем мозге несколько протеопатий могут перекрываться. Например, в дополнение к таупатии и Aβ-амилоидозу (которые сосуществуют как ключевые патологические признаки болезни Альцгеймера), многие пациенты с болезнью Альцгеймера имеют сопутствующую синуклеинопатию (тельца Леви ) в мозге <. 135>
Предполагается, что шапероны и ко-шапероны (белки, которые помогают сворачиванию белка ) могут противодействовать протеотоксичности во время старения и при заболеваниях, связанных с неправильным сворачиванием белков, для поддержания протеостаза..
Некоторые белки могут быть индуцированы к формированию аномальных сборок путем воздействия того же (или аналогичного) белка, который свернулся в вызывающую заболевание конформацию, процесс, называемый «посев» или «разрешающий шаблон» '. Таким образом, болезненное состояние может быть вызвано у восприимчивого хозяина путем введения экстракта пораженной ткани от пораженного донора. Наиболее известной формой такой индуцибельной протеопатии является прионная болезнь, которая может передаваться при воздействии на организм хозяина очищенного прионного белка в вызывающей заболевание конформации. В настоящее время имеются доказательства того, что другие протеопатии могут быть вызваны аналогичным механизмом, включая амилоидоз Aβ, амилоидоз амилоида A (AA) и амилоидоз аполипопротеина AII, таупатию, синуклеинопатию и агрегацию супероксиддисмутазы -1 (SOD1), полиглутамина и ДНК-связывающего белка TAR-43 (TDP-43 ).
Во всех этих случаях аберрантная форма самого белка, по-видимому, быть патогенным агентом. В некоторых случаях отложение одного типа белка может быть экспериментально вызвано агрегированными ансамблями других белков, которые имеют богатую структуру β-слоя, возможно, из-за структурной комплементарности белковых молекул. Например, амилоидоз AA может стимулироваться у мышей такими разнообразными макромолекулами, такими как шелк, дрожжевой амилоид Sup35 и фибриллы курли из бактерии Escherichia coli. Кроме того, амилоид аполипопротеина AII может быть индуцирован у мышей с помощью множества богатых β-листами амилоидных фибрилл. ls, а церебральная таупатия может быть вызвана экстрактами мозга, которые богаты агрегированным Aβ. Имеются также экспериментальные данные о перекрестном посеве между прионным белком и Aβ. В общем, такой гетерологичный посев менее эффективен, чем посев поврежденной формой того же самого белка.
