
Войти
| Дефицит мевалонаткиназы | |
|---|---|
| Другие имена | Мевалоновая ацидурия и синдром гипер иммуноглобулина D (HIDS) |
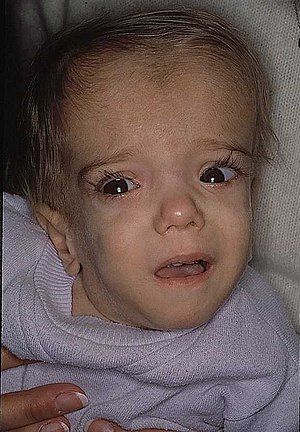 | |
| Пациент с дефицитом мевалонаткиназы в возрасте 21 месяца с характерными черепно-лицевыми особенностями. | |
| Специальность | Гематология, неврология, иммунология, медицинская генетика, эндокринология |
 Мевалоновая кислота
Мевалоновая кислота Мевалонат киназа дефицит ( МКД), является аутосомно - рецессивным нарушением обмена веществ, что приводит к нарушению биосинтез из холестерина и изопреноидов. Это очень редкое генетическое заболевание.
Для него характерен повышенный уровень иммуноглобулина D в крови. Мевалонаткиназа (MVK) - это фермент, участвующий в биосинтезе холестеринов и изопреноидов, и он необходим для превращения мевалоната в мевалонат-5-фосфат в присутствии Mg 2+. MKD возникает из-за мутации в гене, кодирующем мевалонаткиназу, что приводит к снижению или недостаточной активности этого фермента. Из-за этого дефицита мевалоновая кислота может накапливаться в организме, причем высокие уровни обнаруживаются в моче. Тяжесть МКБ зависит от уровня этого дефицита, при этом синдром гипериммуноглобулинемии D (впервые описанный как HIDS в 1984 г.) является менее тяжелым, но более распространенным, а также мевалоновой ацидурией (MVA); более тяжелая, но более редкая форма.
Дефицит мевалонаткиназы наследуется по аутосомно-рецессивному типу, что означает, что ребенок должен унаследовать дефектную копию гена от обоих родителей, чтобы пострадать. Это пример мутации с потерей функции. Ген, кодирующий мевалонаткиназу, состоит из 10 экзонов в локусе 12q14. Было охарактеризовано около 63 патологических вариантов последовательности гена. Наиболее распространенными из них являются V377I, I268T, H20P / N и P167L, которые присутствуют у 70% больных.
Иммуноглобулин D (IgD) - это белок, вырабатываемый определенным типом белых кровяных телец. Есть пять классов иммуноглобулинов : IgG, IgA, IgM, IgE и IgD. Каждый из них играет важную роль в иммунной системе. Функция IgD до сих пор неясна, хотя одним из многих его эффектов является активация иммунной системы.
 Биосинтез изопреноидов
Биосинтез изопреноидов Существует повышенная секреция лихорадки, способствующая развитию цитокина интерлейкина 1 бета (IL-1β) в МКД, что, скорее всего, опосредовано пренилированием дефектного белка. Пренилирование относится к добавлению к белкам гидрофобных изопреноидов, таких как фарнезилпирофосфат (FPP) или геранилгеранилпирофосфат (GGPP). Когда изопреноиды, подобные этим, соединяются с целевым белком, это влияет на расположение и функцию белка в клетке. На модели моноцитарной МКД человека было обнаружено, что дефицит GGPP приводит к избыточной продукции IL-1β и дефектному пренилированию RhoA. Это вызывает повышенный уровень Rac1 и PKB, что, в свою очередь, влияет на GTPases и B7-гликопротеины. Ранее было обнаружено, что путь Rac1 / PI3K / PKB был связан с патогенезом MKD. Инактивация RhoA действует как индуктор транскрипции мРНК IL-1β независимо от активности NLRP3 или каспазы-1. Из-за дефектного RhoA в клетке образуются дефектные митохондрии (удлиненные и нестабильные). Обычно дефектные митохондрии очищаются в клетке за счет механизма аутофагии. Но при МКД нарушается клиренс дефектных митохондрий из цитозоля. В результате митохондриальная ДНК начинает накапливаться в цитозоле, связывая и активируя NLRP3, который отвечает за выработку IL-1β. Активация может быть прямой или косвенной. Он также может быть активирован реактивными формами кислорода (АФК). Известно, что моноциты и макрофаги у пораженных людей также продуцируют более высокие уровни фактора некроза опухоли альфа ( TNF-α ), интерлейкина 6 ( IL-6 ), отличного от IL-Iβ. Во время приступов лихорадки (лихорадки), C-реактивного белка ( CRP) ) также увеличивается. СРБ выделяется печенью, что вызывает воспаление.
Гипериммуноглобулинемия D с рецидивирующей лихорадкой - это синдром периодической лихорадки, первоначально описанный в 1984 году терапевтом Йосом ван дер Меером, затем в Медицинском центре Лейденского университета. В мире описано не более 300 случаев. В настоящее время он признан аллельным вариантом MKD.
HIDS - один из множества синдромов периодической лихорадки. Для него характерны приступы лихорадки, артралгии, поражения кожи, включая циклические язвы во рту, и диарею. Лабораторные признаки включают острофазовый ответ (повышенный СРБ и СОЭ ) и заметно повышенный IgD (и часто IgA ), хотя описаны случаи с нормальным IgD.
В основном он был описан в Нидерландах и Франции, хотя в международный регистр включен ряд случаев из других стран.
Дифференциальный диагноз включает лихорадка неизвестного происхождения, семейной средиземноморской лихорадки (ССЛ) и семейная Хиберниан лихорадка (или ФНО прием связан периодический синдром / ловушки).
Практически у всех людей с синдромом имеют мутации в гене для мевалоната киназы, которая является частью редуктазы пути HMG-CoA, что является важным клеточного метаболизма. Действительно, подобные приступы лихорадки (но с нормальным уровнем IgD) были описаны у пациентов с мевалоновой ацидурией - врожденной ошибкой метаболизма, которая теперь рассматривается как тяжелая форма HIDS.
Неизвестно, как мутации мевалонаткиназы вызывают приступы лихорадки, хотя предполагается, что другие продукты пути биосинтеза холестерина, цепи пренилирования ( геранилгераниол и фарнезол ) могут играть роль.
Дефицит мевалонаткиназы вызывает накопление мевалоновой кислоты в моче в результате недостаточной активности фермента мевалонаткиназы (АТФ: мевалонат-5-фосфотрансфераза; EC 2.7.1.36).
 Мевалонатный путь
Мевалонатный путь Расстройство было впервые описано в 1985 году.
Дефицит мевалонаткиназы, классифицируемый как врожденная ошибка метаболизма, обычно приводит к задержке развития, гипотонии, анемии, гепатоспленомегалии, различным дисморфическим особенностям, умственной отсталости, общей неспособности к развитию и ряду других признаков.
 Дефицит мевалонаткиназы имеет аутосомно-рецессивный характер наследования.
Дефицит мевалонаткиназы имеет аутосомно-рецессивный характер наследования. МКД не лечится. Но воспаление и другие эффекты можно до некоторой степени уменьшить.
Канакинумаб был одобрен для лечения HIDS и показал свою эффективность. Иммунодепрессанты этанерцепт и Anakinra также показали, чтобы быть эффективными. Статиновые препараты могут снижать уровень мевалоната, и в настоящее время они исследуются. В недавнем сообщении об одном случае бисфосфонаты были выделены как потенциальный терапевтический вариант.
Во всем мире менее 1 человека из 100 000 страдают HIDS, из них около 200 человек страдают MKD. Это классифицирует состояние как редкое генетическое заболевание.
| Классификация | D |
|---|---|
| Внешние ресурсы |
| Классификация | D |
|---|