
Войти
| Лейкодистрофия | |
|---|---|
 | |
| Т2-взвешенная аксиальная сканирование на уровне хвостатого головы демонстрирует заметную потерю заднего белого вещества с уменьшенным объемом и повышенной интенсивностью сигнала. Переднее белое вещество сохраняется. Характеристики соответствуют Х-связанной адренолейкодистрофии. | |
| Специальность | Неврология |
Лейкодистрофии - это группа обычно наследуемых заболеваний, характеризующихся дегенерацией белое вещество в мозге. Слово лейкодистрофия происходит от греческого корня leuko, «белый», dys, «ненормальный» и troph, «рост». Лейкодистрофии вызываются несовершенным ростом или развитием миелиновой оболочки, жировой изолирующей оболочки вокруг нервных волокон. Лейкодистрофии можно классифицировать как гипомиелинизирующие или демиелинизирующие заболевания, в зависимости от того, присутствует ли повреждение до рождения или возникает после него. Другие демиелинизирующие заболевания обычно не являются врожденными и имеют токсическую или аутоиммунную причину.
Когда происходит повреждение белого вещества, иммунные реакции могут привести к воспалению в центральной нервной системе (ЦНС), а также с потерей миелина. Дегенерацию белого вещества можно увидеть на МРТ и использовать для диагностики лейкодистрофии. Лейкодистрофия характеризуется специфическими симптомами, включая снижение двигательной функции, ригидность мышц и возможное ухудшение зрения и слуха. Хотя заболевание является смертельным, возраст начала является ключевым фактором, поскольку средняя продолжительность жизни младенцев составляет 2–8 лет, в то время как взрослые обычно живут более десяти лет после начала. Варианты лечения ограничены, хотя трансплантация гемопоэтических стволовых клеток с использованием костного мозга или пуповинной крови, похоже, помогает при определенных типах, пока проводятся дальнейшие исследования.
Общая частота лейкодистрофий оценивается в 1 из 7600. Большинство типов связано с наследованием Х-сцепленного рецессивного или Х-сцепленного доминантного признака, в то время как другие, хотя и включают дефектный ген, являются результатом спонтанного мутация, а не генетическая наследственность.
Некоторые специфические симптомы варьируются от одного типа лейкодистрофии к другому, но подавляющее большинство симптомов общие причины болезни обычно имеют те же последствия. Симптомы зависят от возраста начала, который преимущественно приходится на младенчество и раннее детство, хотя точное время появления может быть трудно определить. Повышенная раздражительность и гиперчувствительность к окружающей среде являются обычными явлениями, а также некоторые контрольные физические признаки, включая жесткость мышц и запрокинутую голову. Ботокс-терапию часто используют для лечения пациентов со спастичностью. У детей и взрослых проявляются похожие симптомы, включая снижение или потерю слуха и зрения. Хотя у детей действительно наблюдается дегенерация зрительного и слухового аппарата, течение болезни обычно слишком быстрое, что приводит к относительно быстрой смерти, тогда как взрослые могут жить с этими состояниями в течение многих лет. У детей спастическая активность часто предшествует прогрессирующей атаксии и быстрому когнитивному ухудшению, которое было описано как умственная отсталость. Эпилепсия является обычным явлением для пациентов любого возраста. У более прогрессирующих пациентов наблюдается слабость глотания, что приводит к приступам спастического кашля из-за вдыхания слюны. Классическое симптоматическое прогрессирование ювенильной Х-сцепленной адренолейкодистрофии показано в фильме 1992 г. Масло Лоренцо.
Курс и расписание зависят от возраста начала, а младенцы демонстрируют продолжительность жизни 2–8 лет., подростки 2–10 лет и взрослые обычно 10+ лет. Взрослые обычно наблюдают длительный период стабильности, за которым следует снижение до вегетативного состояния и смерть. Хотя методы лечения действительно существуют, большинство из них находятся в экспериментальной фазе и могут обещать только остановку прогрессирования симптомов, хотя некоторые генные методы лечения показали некоторое улучшение симптомов. Изнурительное течение болезни привело к многочисленным философским и этическим аргументам по поводу экспериментальных клинических испытаний, прав пациентов и самоубийства с помощью врача.
В то время как более конкретные основные причины лейкодистрофии зависят от в зависимости от типа, однако, существуют общие патофизиологические закономерности, которые можно увидеть среди всех типов. Прежде всего, лейкодистрофия - это нейродегенеративное заболевание, которое всегда является результатом как нарушения, так и поддержания миелиновых оболочек, окружающих нейронные аксоны в центральной нервной системе в качестве основного результат генетической мутации. Миелин представляет собой жирное белое вещество, которое действует как электрический изолятор и покрывает аксоны, чтобы ускорить импульсы (т.е. потенциалы действия ), идущие вниз по аксону. Таким образом, естественным результатом потери этого вещества является снижение эффективности распространения импульсов. Поскольку миелин вырабатывается олигодендроцитами (тип глиальных клеток ) в центральной нервной системе, проще всего искать причину в мутации или неисправности этих клеток и в других глиальных клетках.
 Модель ауторецессивного наследования
Модель ауторецессивного наследования Лейкодистрофия чаще всего является наследственным заболеванием, которое обычно является результатом аутосомно-рецессивной модели наследования, хотя доминантные модели наследования не являются чем-то необычным, как в случае лейкодистрофии у взрослых. Это означает, что затронутый аллель находится на аутосомной или неполовой хромосоме и маскируется доминантным, незатронутым фенотипом. Другими словами, чтобы индивид унаследовал фенотип лейкодистрофии, он или она должны иметь два рецессивных мутантных аллеля. Болезнь Краббе и метахроматическая лейкодистрофия (MLD) - два таких типа. MLD обнаруживается на хромосоме 22 человека в положении q13.31. Другой тип наследственной лейкодистрофии - это Х-сцепленная адренолейкодистрофия (X-ALD). Как следует из названия, этот тип лейкодистрофии является результатом мутации, обнаруженной на Х-хромосоме. Он также носит рецессивный характер. Х-хромосома - это половая хромосома, и поскольку у женщин есть два «шанса» получить нормальную Х-хромосому (одна материнская, одна отцовская), а у мужчин только одна (одна материнская), это заболевание более вероятно. быть замеченным у мужчин, чем у женщин. Мутация, приводящая к лейкодистрофии у взрослых, обозначена как 5q23.
Несмотря на то, что существует около сорока различных типов лейкодистрофий, многие из них отсутствуют в формальных и всесторонних исследованиях. На данный момент большая часть исследований была проведена по пяти типам: (1) метахроматическая лейкодистрофия (MLD), (2) болезнь Краббе, (3) X-Linked адренолейкодистрофия (ALD), (4) болезнь Канавана и (5) болезнь Александера. Каждый тип лейкодистрофии имеет уникальную патофизиологию, но все пять из них тем или иным образом влияют на подмножество глиальных клеток, нарушая выработку и поддержание миелина, и обычно связаны с мутациями с участием генов, кодирующих ферменты, необходимые для катаболизм жирных кислот с очень длинной цепью (ЖКОДЦ), токсичных для миелин-продуцирующих клеток центральной нервной системы.
Метахроматическая лейкодистрофия - это результат генетических дефектов ферментов, связанных с клеточным компартментом лизосомой. MLD является одним из двух лейкодистофий, которые также являются лизосомным расстройством накопления. MLD наследуется аутосомно-рецессивным способом и является результатом мутаций в трех разных аллелях ARSA , которые кодируют фермент арилсульфатазу A (ASA или иногда ARSA), также называется сульфатид сульфатаза. ASA отвечает за распад сульфатидов, сфинголипидов,, присутствующих в мембранах нейронов, а также в миелине. Когда есть мутация в гене, кодирующем ASA, результатом является снижение продукции, что впоследствии приводит к уменьшению разложения сульфатидов, вызывая их накопление. Это накопление сульфатидов ядовито для олигодендроцитов, миелин-продуцирующих клеток ЦНС, эффективно приводя к нарушению структуры миелина с последующим демиелинизацией. Тип наследования трех разных аллелей влияет на то, какой тип MLD у человека развивается. Два нулевых аллеля отвечают за инфантильную версию и не позволяют продуцировать ASA. У гетерозиготного индивидуума (один нулевой аллель, один ненулевой аллель) развивается ювенильная форма и наблюдается некоторая продукция ASA, в то время как индивидуум с двумя ненулевыми аллелями (но все еще мутировавший) развивает взрослую форму.
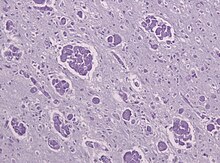 Лейкодистрофия глобоидных клеток PAS - Многоядерные макрофаги («глобоидные клетки») и потеря миелинизированных волокон в случае лейкодистрофии Краббе
Лейкодистрофия глобоидных клеток PAS - Многоядерные макрофаги («глобоидные клетки») и потеря миелинизированных волокон в случае лейкодистрофии Краббе Подобно MLD, болезнь Краббе является другой тип лейкодистрофии с аутосомно-рецессивным наследованием, который является результатом лизосомного расстройства накопления. Это происходит из-за делеции в экзоне 16 гена GALC, которая вызывает мутацию сдвига рамки считывания, приводящую к преждевременному возникновению стоп-кодона. Ген GALC, обнаруженный на хромосоме 14 в положении 31 (14q31), кодирует фермент бета-галактоцереброзидазу (GALC). GALC - это лизосомальный фермент, ответственный за катаболизм галактолипидов, особенно психозина, которые широко распределяются по всему мозгу. Таким образом, дефицит GALC вызывает накопление этих жирных кислот, известных как глобоидные макрофаги, которые разрушают олигодендроциты, тем самым подавляя образование миелина.
Из-за присутствия глобоидных клеток. сгруппированная около белого вещества, болезнь Краббе часто называется лейкодистрофией глобоидных клеток. Кроме того, новое исследование показало, что болезнь Краббе и лейкодистрофия глобоидных клеток могут быть разными заболеваниями из-за секреции медиаторов воспаления в некоторых случаях естественными клетками-киллерами. Это исследование показало, что клетки Natural Killer имеют рецепторы (TDAG8) для определенных гликосфинголипидов, которые накапливаются у человека с лейкодистрофией, опять же из-за недостаточного уровня GALC, и, будучи связанными, нацелены на клетки Natural Killer для разрушения, тем самым предотвращение их цитотоксических эффектов. Эти сфинголипиды были идентифицированы как галактозил-сфингозин и гликозил-сфингозин и не присутствуют у здоровых людей.
Болезнь Канавана является менее изученным типом лейкодистрофии, который, как и MLD и болезнь Краббе, также передается по типу аутосомно-рецессивного наследования. Это связано с мутацией в гене ASPA, который кодирует аспартоацилазу, фермент, необходимый для метаболизма N-ацетил-L-аспартата (NAA). Мутация вызывает дефицит аспартоциклазы. NAA участвует в образовании липидов, и если она не расщепляется аспартоацилазой, ее избыточные уровни накапливаются, вызывая демиелинизацию.
При X-связанной адренолейкодистрофии (X-ALD) мутация происходит в пероксисомальной АТФ-связывающей кассете (переносчик ABC ). Это приводит к воспалительной демиелинизации мозга, вызванной дестабилизацией миелина, которая происходит у этих пациентов. Воспалительная демиелинизация начинается в мозолистом теле и медленно прогрессирует к обоим полушариям. У пациентов с X-ALD аномально высокие уровни очень длинноцепочечных жирных кислот (VLCFA) накапливаются в различных тканях и жидкостях организма. Эта повышенная концентрация затем включается в различные сложные липиды, где они обычно не встречаются. Было обнаружено, что это непосредственно связано с церебральным воспалением. Накопленные и встроенные ЖКОДЦ в сложные липиды могут привести к дестабилизации миелиновой оболочки и, в конечном итоге, к демиелинизации.
Болезнь Александера отличается от лейкодистрофий, упомянутых выше, тем, что она является результат спонтанной мутации, то есть не передается по наследству. Это означает, что мутация, обнаруженная у пораженного человека, не обнаружена ни у одного из его родителей. Это связано с накоплением глиального фибриллярно-кислого белка (GFAP ) в результате мутации в гене GFAP; который, вместо того, чтобы быть обнаруженным в ассоциации с лизосомами или пероксисомами, представляет собой промежуточный филамент, связанный с ядерной оболочкой. Промежуточные филаменты - это белки, ответственные за структуру клеточного цитоскелета, и, таким образом, этот тип мутации участвует в нарушении структурного развития клеток. Фактически, дефекты цитоскелета и молекулы-переносчика наблюдались в астроцитах (тип глиальных клеток) пораженных людей. Эти астроциты содержат нездорово большое количество GFAP, которое влияет на формирование и функцию астроцитов.
Дегенерация белого вещества, которая показывает дегенерацию миелина можно увидеть на простом МРТ и использовать его для диагностики лейкодистрофий всех типов. Наиболее полезными являются взвешенные изображения T-1 и T-2 FLAIR. FLAIR означает инверсионное извлечение с ослаблением флюида. Также могут проводиться электрофизиологические и другие виды лабораторных исследований. В частности, скорость нервной проводимости исследуется, чтобы различать лейкодистрофию и другие демиелинизирующие заболевания, а также различать отдельные лейкодистрофии. Например, люди с X-ALD имеют нормальную скорость проводимости, в то время как люди с болезнью Краббе или метахроматической лейкодистрофией имеют отклонения в скорости проводимости. Панели мультигенного секвенирования нового поколения для недифференцированной лейкодистрофии теперь могут быть предложены для быстрой молекулярной диагностики после соответствующего генетического консультирования.
Конкретные типы лейкодистрофий включают следующие с соответствующими МКБ-10 коды, при их наличии:
При множестве различных типов лейкодистрофий и причин лечебная терапия различается для каждого типа. Многие исследования и клинические испытания находятся в стадии разработки, чтобы найти лечение и терапию для каждой из различных лейкодистрофий. Трансплантаты стволовых клеток и генная терапия представляются наиболее перспективными в лечении всех лейкодистрофий при условии, что это делается как можно раньше. В отношении гипомиелинизирующих лейкодистрофий терапевтические исследования клеточной терапии представляются многообещающими. Клетки-предшественники олигодендроцитов и нервные стволовые клетки были успешно трансплантированы и год спустя оказались здоровыми. Карты фракционной анизотропии и радиальной диффузии показали возможную миелинизацию в области трансплантата. Индуцированные плюрипотентные стволовые клетки, клетки-предшественники олигодендроцитов, генная коррекция и трансплантация для ускорения созревания, выживания и миелинизация олигодендроцитов, по-видимому, является основным путем возможного лечения.
Для трех типов лейкодистрофий (X-сцепленная адренолейкодистрофия (X-ALD), метахроматическая лейкодистрофия (MLD) и болезнь Краббе (глобоидноклеточная лейкодистрофия - GLD), генная терапия с использованием аутологичных гемопоэтических стволовых клеток для переноса гена заболевания с лентивирусом векторы оказались успешными и в настоящее время используются в клинических испытаниях для X-ALD и MLD. Прогрессирование X-ALD было нарушено генной терапией гемопоэтическими стволовыми клетками, но точная причина, почему демиелинизация останавливается, и количество необходимых стволовых клеток неясно. представляет собой накопление жирных кислот с очень длинной цепью в головном мозге, это не похоже на причину заболевания, поскольку генная терапия не исправляет его.
Аденоассоциированные векторы также использовались во внутримозговых инъекциях для лечения MLD. У некоторых пациентов с MLD их IQ увеличился, нервная проводимость улучшилась, их МРТ выглядели стабильными и имели нормальный уровень ферментов. Хотя у подавляющего большинства пациентов после трансплантации состояние улучшается, некоторые плохо реагируют на лечение, что может привести к разрушительным результатам. Для тех лейкодистрофий, которые возникают в результате дефицита лизоцимных ферментов, таких как болезнь Краббе, заместительная ферментная терапия кажется обнадеживающей. Однако доставка ферментов оказывается сложной, потому что гематоэнцефалический барьер сильно ограничивает то, что может проходить в центральную нервную систему. Из-за этого препятствия большинство исследований и клинических испытаний обращаются к аллогенной трансплантации гемопоэтических стволовых клеток.
 Х-сцепленное рецессивное наследование
Х-сцепленное рецессивное наследование В настоящее время ни одно исследование не показало более высокую распространенность большинства типов лейкодситрофии. в любом месте по всему миру. Однако существует более высокая распространенность болезни Канавана среди еврейского населения. Каждый 40 человек ашкенази еврейского происхождения является носителем болезни Канавана. Это оценивается примерно в 2,5%. Кроме того, из-за паттернов аутосомно-рецессивного наследования не обнаружено значительных различий между пораженными мужчинами и пораженными женщинами для большинства типов лейкодистрофии, включая, помимо прочего, метахроматическую лейкодистрофию, болезнь Краббе, болезнь Канавана и болезнь Александра. Единственным исключением из этого правила является любой тип лейкодистрофии, переносимой на половой хромосоме, например, Х-сцепленная адренолейкодистрофия, переносимая на Х-хромосоме. Из-за наследственности Х-сцепленных заболеваний мужчины чаще страдают этим типом лейкодистрофии, хотя у женщин-носителей часто наблюдаются симптомы, хотя и не так сильно, как у мужчин. На сегодняшний день не было обнаружено случаев лейкодистрофии, переносимой на Y-хромосому.
Кроме того, многие исследовательские группы изучают клеточные процессы миелинизации, что может дать понимание в лейкодистрофию. Исследователи из Нью-Йорка успешно вылечили лейкодистрофию у мышей, используя клетки кожи для восстановления поврежденных миелиновых оболочек. Исследователи предполагают, что это лечение может быть использовано для лечения рассеянного склероза у человека.
United Leukodystrophy Foundation (ULF), зарегистрированная в 1982 году, является некоммерческой организацией., добровольная медицинская организация, занимающаяся финансированием передовых исследований и предоставлением пациентам и их семьям информации о заболеваниях и направлений к врачам.
Cure MLD - это глобальная сеть защитников интересов пациентов и некоммерческих организаций, занимающихся оказанием помощи семьям, пострадавшим от метахроматической лейкодистрофии (MLD). Cure MLD здесь, чтобы соединить семьи с ресурсами, информацией, поддержкой и другими лицами, имеющими отношение к MLD. Cure MLD предоставляет гранты на поездки, пакеты услуг, эмоциональную поддержку, образовательные материалы и возможности для общения с семьями и пациентами. Поддержку сайту оказывают Chloe's Fight, Love for Loie and the Hammond Family, The Sullivan Family and Friends, Gavin Flying for a Cure, The Calliope Joy Foundation / Cal's Cupcakes, Fundacion Lautaro te Necesita (Аргентина), Ресурсная и исследовательская организация по лейкодистрофии. (Австралия), Bethany's Hope (Канада) и семьи MLD по всему миру. "Мы едины в нашей миссии: Cure MLD "
MLD Foundation был основан Дином и Терин Зур в 2001 году после того, как в 1995 году двум их дочерям был поставлен диагноз MLD. MLD Foundation обслуживает семьи и работает с исследователями, клиницистами, регулирующими органами, плательщиками и политиками по всему миру по вопросам MLD, лейкодистрофии, лизосомных заболеваний и редких заболеваний. Миссия фонда - «We CARE ™... C безумие для семей, повышая A осведомленность, влияя и финансируя R исследования и продвигая E дуктацию от метахроматической лейкодистрофии, очень редкого терминального генетического нейрометаболического заболевания, у которого более половины
Всемирный лейкодистрофический альянс повышает осведомленность и работает над улучшением качества лечения лейкодистрофий.
Джилл Келли и ее муж, НФЛ защитник Джим Келли, основанный Hunter's Hope после того, как их сыну Хантеру (1997-2005) поставили диагноз инфантильный e Лейкодистрофия Краббе. Мэтью и Майкл Кларк из Hull, Великобритания пострадали, к сожалению, оба скончались в 2013 и 2016 годах соответственно. Их история стала предметом документального фильма Channel 4 «Загадочная история братьев Кларк».
Августо и Микаэла Одоне основали The Myelin Project после того, как их сын, Лоренцо был поставлен диагноз адренолейкодистрофия (АБП). Фильм 1992 года Масло Лоренцо - это правдивая история о мальчике, страдающем адренолейкодистрофией (ALD).
| Классификация | D |
|---|