Войти
Аденинфосфорибозилтрансфераза (APRTase ) - это фермент, кодируемый геном APRT , обнаруженный у людей на хромосоме 16. Он является частью семейства PRTase типа I и участвует в пути спасения нуклеотидов, который обеспечивает альтернативу биосинтезу нуклеотидов de novo у людей и большинства других животных. У паразитарных простейших, таких как лямблии, APRTase обеспечивает единственный механизм, с помощью которого может производиться аденин. Дефицит APRTase способствует образованию камней в почках (мочекаменная болезнь ) и потенциальной почечной недостаточности.
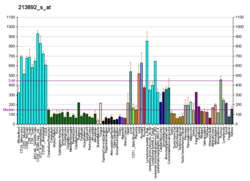
 Ген APRT состоит из 5 экзонов (показаны синим цветом). Стартовый (ATG) и стоповый (TGA) кодоны обозначены жирным синим шрифтом. Динуклеотиды CpG выделены красным цветом. Их больше в вышестоящей области гена, где они образуют островок CpG.
Ген APRT состоит из 5 экзонов (показаны синим цветом). Стартовый (ATG) и стоповый (TGA) кодоны обозначены жирным синим шрифтом. Динуклеотиды CpG выделены красным цветом. Их больше в вышестоящей области гена, где они образуют островок CpG.APRTase катализирует следующую реакцию в пути спасения пуриновых нуклеотидов :
Аденин + фосфорибозилпирофосфат ( PRPP ) → Аденилат (AMP ) + пирофосфат (PPi )
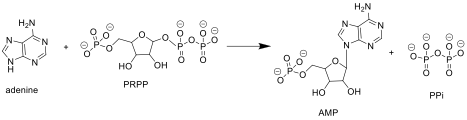 ARPTase катализирует перенос фосфорибозила от PRPP к аденину, образуя AMP и высвобождая пирофосфат (PPi).
ARPTase катализирует перенос фосфорибозила от PRPP к аденину, образуя AMP и высвобождая пирофосфат (PPi). У организмов, которые могут синтезировать пурины de novo, путь спасения нуклеотидов обеспечивает альтернативу, которая является энергетически более эффективной. Он может спасти аденин из пути биосинтеза полиамина или из пищевых источников пуринов.. Хотя APRTase функционально избыточна у этих организмов, она становится более важной в периоды быстрого роста, такие как эмбриогенез и рост опухоли. тыс. Он конститутивно экспрессируется во всех тканях млекопитающих.
У простейших паразитов путь спасения нуклеотидов обеспечивает единственное средство для синтеза нуклеотидов. Поскольку последствия дефицита APRTase у людей сравнительно легкие и поддаются лечению, некоторые паразитарные инфекции можно лечить, воздействуя на функцию APRTase.
В растениях, поскольку у других организмов ARPTase функционирует в основном для синтеза аденилата. Он обладает уникальной способностью метаболизировать цитокинины - гормон растения, который может существовать в виде основания, нуклеотида или нуклеозида. - нуклеотиды аденилата.
APRT функционально родственен гипоксантин-гуанинфосфорибозилтрансферазе (HPRT).
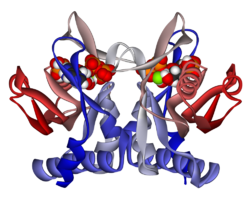
APRTase представляет собой гомодимер со 179 аминокислотными остатками на мономер. Каждый мономер содержит следующие области:
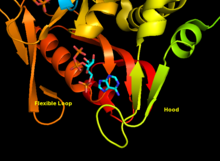 Каталитический сайт APRTase с разделенными реагентами аденином и PRPP. Считается, что «капюшон» важен для пуриновой специфичности, в то время как гибкая петля, как полагают, содержит молекулы в активном сайте.
Каталитический сайт APRTase с разделенными реагентами аденином и PRPP. Считается, что «капюшон» важен для пуриновой специфичности, в то время как гибкая петля, как полагают, содержит молекулы в активном сайте. 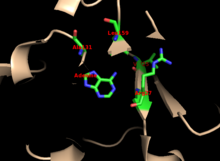 Остатки A131, L159, V25 и R27 важны для пуриновой специфичности в человеческой APRTase.
Остатки A131, L159, V25 и R27 важны для пуриновой специфичности в человеческой APRTase. Ядро высоко консервативно для многих PRTase. Капюшон, содержащий сайт связывания аденина , имеет большую вариабельность в семействе ферментов. Мотив из 13 остатков включает связывающую область PRPP и включает два соседних кислотных остатка и по крайней мере один окружающий гидрофобный остаток.
специфичность к аденину включает гидрофобные остатки Ala131 и Leu159 в коровом домене. У людей два остатка в капюшоне водородной связи с пурином для дополнительной специфичности: Val25 с атомами водорода на N6 и Arg27 с N1. Хотя гибкая петля не взаимодействует с капюшоном во время распознавания пурина, считается, что она закрывает активный сайт и изолирует реакцию растворителей.
Большинство исследований APRTase сообщает, что Mg имеет важное значение. для переноса фосфорибозила, и это сохраняется для PRTаз типа I. Однако недавняя попытка разрешить структуру APRTase человека не смогла найти единственный сайт для Mg, но обнаружила доказательства, позволяющие предположить наличие атома Cl рядом с Trp98. Несмотря на сложность размещения Mg, общепринято считать, что каталитический механизм зависит от этого иона.
APRTase действует по двунаправленному последовательному механизму, с образованием тройного комплекса. Фермент сначала связывает PRPP, а затем аденин. После того, как происходит перенос фосфорибозила, первым уходит пирофосфат, а затем АМФ. Кинетические исследования показывают, что перенос фосфорибозила происходит относительно быстро, в то время как высвобождение продукта (особенно высвобождение АМФ) ограничивает скорость.
Считается, что в APRTase человека протон N9 аденина отводится Glu104 для образования оксакарбениевого переходного состояния. Он действует как нуклеофил, чтобы атаковать аномерный углерод PRPP, образуя AMP и вытесняя пирофосфат из PRPP. Механизм APRTase в целом согласуется с механизмом других PRTases, которые сохраняют функцию замещения α-1-пирофосфата PRPP с использованием нуклеофила азота либо в S N 1, либо в S Атака N 2.
Когда APRTase имеет сниженную или отсутствующую активность, аденин накапливается из других путей. Он разлагается ксантиндегидрогеназой до 2,8-дигидроксиаденина (DHA). Хотя DHA связана с белками плазмы, она имеет плохую растворимость в моче и постепенно осаждается в почечных канальцах, что приводит к образованию камней в почках (мочекаменная болезнь ). Если не лечить, это состояние может в конечном итоге привести к почечной недостаточности..
Дефицит ARPTase был впервые диагностирован в UK в 1976 году. С тех пор у людей были определены две категории дефицита APRTase.
Дефицит типа I приводит к полной потере активности APRTase и может возникать у пациентов, гомозиготных или сложных гетерозиготных по различным мутациям. Секвенирование выявило множество различных мутаций, которые могут составлять тип 1, включая миссенс-мутации, бессмысленные мутации, дублированный набор из 4 пар оснований в экзон 3 и единственная вставка тимина в интрон 4. Эти мутации вызывают эффекты, которые сгруппированы в три основных области: в связывании β-фосфата PRPP, в связывании 5'-фосфата PRPP и в сегменте гибкой петли, которая закрывает активный сайт во время катализа дефицита типа I. наблюдался у различных этнических групп, но изучался преимущественно среди популяций белых.
Дефицит типа II вызывает снижение сродства APRTase к PRPP, что приводит к десятикратному увеличению K Значение M. Это наблюдалось и изучалось в основном в Японии.
. Диагноз дефицита APRTase может быть поставлен путем анализа камней в почках, измерения концентрации DHA в моче или анализа активности APRTase в эритроцитах. Его можно лечить с помощью обычных доз аллопуринола или фебуксостата, которые ингибируют активность ксантиндегидрогеназы для предотвращения накопления и осаждения DHA. Состояние также можно уменьшить с помощью диеты с низким содержанием пуринов и большого количества жидкости.




 ..
..