
Войти
| Семейный названия аденоматозный полипоз | |
|---|---|
| Другие названия | FAP |
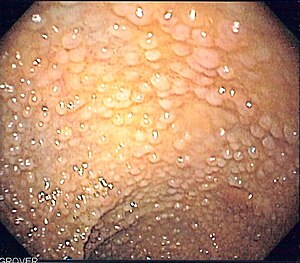 | |
| Эндоскопическое изображение сигмовидная кишка пациент с семейным аденоматозным полипозом | |
| Специальность | Гастроэнтерология, Онкология |
| Начало Осложнения | Колоректальный рак |
| Обычное | <35 years of age |
| Продолжительность | Пожизненный |
| Типы | Классический или ослабленный |
| Вызывает | мутация гена APC |
| Метод диагностики | Колоноскопия. Генетическое тестирование |
| Дифференциальный диагноз | синдром Линча, MUTYH-ассоциированный полипоз |
| Лечение | Колоноскопия. Полипэктомия. Верхняя эндоскопия. Колэктомия |
| Частота | 1 из 10,000 - 15,000 |
Семейный аденоматозный полипоз (ФАП ) - это аутосомно-доминантное наследственное заболевание, при котором многочислен аденоматозные полип ы образуются в основном в эпителии толстой кишки. Хотя эти полипы начинаются доброкачественными, злокачественная трансформация в рак толстой кишки происходит, когда их не лечить. Известно три варианта: FAP и аттенуированный FAP (используемый называвшийся синдром наследственной плоской аденомы ) вызваны дефекта гена APC на хромосоме 5, тогда как аутосомно-рецессивный FAP (или MUTYH-ассоциированный полипоз ) вызывается дефектами гена MUTYH на хромосоме 1. Из этих трех сам FAP является самыми тяжелыми и самыми распространенными; хотя для всех трех образующиеся полипы и рак толстой кишки изначально ограничиваются стенкой толстой кишки. Обнаружение и удаление до метастазов вне толстой кишки может снизить и во многих случаях устранить распространение рака.
Подразумевается, что первопричиной FAP является генетическая мутация - изменение генов-супрессоров опухоли в организме, которые предотвращают развитие опухолей. Это изменение позволяет многочисленным клеткам кишечника развиваться в других раковых полипах, когда они обычно достигают конца своей жизни; неизбежно один или несколько из них в итоге будут прогрессировать и вызвать рак (риск 7% к 21 году, возрастает до 87% к 45 годам и 93% к 50 годам). Эти изменения, генов не вызывает рак, а, скорее, способность организма предотвращать превращение клеток в раковые. Даже с изменением гена может пройти время, прежде чем действительно разовьется клетка, которая в результате становится злокачественной, и в некоторых случаях ген может все еще работать, чтобы контролировать опухоли, поэтому для развития рака FAP требуется много лет, и он почти всегда болезнь, начинающаяся у взрослых.
Вторая форма FAP, известная как аттенуированный семейный аденоматозный полипоз, имеет функциональный ген APC, но несколько нарушен. Таким образом, он может работать как обычно. Ослабленный FAP по-прежнему представляет собой высокий 70% риск развития рака в течение жизни (по оценкам), но обычно проявляется меньшим количеством полипов (обычно 30), чем сотни или тысячи, обычно встречающиеся при FAP, и возникает в возрасте, когда FAP обычно больше не считается вероятным - обычно в возрасте от 40 до 70 лет (в среднем 55), а не более обычных 30 лет и старше. Варианты лечения могут быть разными.
Третий вариант, аутосомно-рецессивный семейный аденоматозный полипоз или MUTYH-ассоциированный полипоз, также более мягкий. и, как следует из названия, требует, чтобы оба родителя были «носителями» для проявления заболевания.
В некоторых случаях ФАП может проявляться выше в толстой кишке, чем обычно (например, восходящая кишка, или проксимальнее изгиба селезенки, или в желудке или двенадцатиперстная кишка), где они не проявляют никаких симптомов до тех пор, пока рак не станет широко распространенным. Мутации APC связаны с другими видами рака, такими как рак щитовидной железы. Мутация, вызывающая FAP, является аутосомно-доминантной, она может быть унаследована напрямую от любого родителя к ребенку. Существует генетический анализ крови гена APC, который может определить, присутствует ли он, и, следовательно, может предсказать возможность FAP. Людям из группы риска (из-за семейных связей или генетического тестирования) обычно предлагается рутинный мониторинг кишечного тракта каждые 1-3 года в течение всей жизни, начиная с полового созревания для FAP и в раннем взрослом возрасте для аттенуированных форм. Операция по резекции толстой кишки рекомендуется, если широко распространены толстой кишки из-за высокого риска ранней смерти от рака толстой кишки. Существуют международные регистры, которые отслеживают известные случаи дефектов генов FAP или APC для исследовательских и клинических целей. Мутация APC также часто встречается в отдельных случаях колоректальной карциномы, что подчеркивает ее важность при этой форме рака.
С раннего подросткового возраста у пациентов с этим заболеванием постепенно (и в большинстве случаев бессимптомно) развиваются от сотен до тысяч колоректальных полипов. (а иногда полипы в других местах ) - небольшие аномалии на поверхности кишечного тракта, особенно толстой кишки, включая толстую кишку или прямая кишка. Они могут кровоточить, что приводит к появлению крови в стуле. Если кровь не видна, у пациента все еще возможно развитие анемии из-за постепенно развивающегося дефицита железа. Если развивается злокачественное новообразование, это может проявляться даже потерей веса, изменением привычки кишечника или метастазами в печень или где-либо еще. ФАП также может развиваться «незаметно» у некоторых людей, проявляет мало или проявляет никаких признаков до тех пор, пока не перерастает в прогрессирующий колоректальный рак.
Потому что семейный полипоз развивается очень постепенно с годами, а также может проявляться в «Ослабленной» формы даже более постепенного развития в результате ФАП, могут привести к развитию рака в любой момент от подросткового до пожилого возраста.
В зависимости от дефекта в гене APC, полной или ослабленной формы, семейный полипоз может проявляться в виде полипов в толстой кишке или в дуоденальном тракте, или в любой их комбинации. Следовательно, отсутствие полипов, например, в прямой кишке, само по себе может быть недостаточным для подтверждения отсутствия полипов. Возможно, потребуется рассмотреть и визуально осмотреть другие возможные части кишечного тракта. Колоноскопия предпочтительнее для этого ректороманоскопии, так как она обеспечивает лучшее наблюдение за обычным правосторонним расположением полипов.
 CHRPE - Врожденная гипертрофия пигментного эпителия сетчатки
CHRPE - Врожденная гипертрофия пигментного эпителия сетчатки Генетическая детерминантаного полипоза также может предрасполагать носителей к другим злокачественным новообразованиям, например, двенадцатиперстной кишки и желудка (особенно ампулярной аденокарциноме). Другими признаками, которые могут указывать на FAP, являются пигментные поражения сетчатки («CHRPE - врожденная гипертрофия пигментного эпителия сетчатки»), кисты челюсти, кисты сальных желез и остеомы. (доброкачественные опухоли костей). Сочетание полипоза, остеом, фибром и кист называется синдромом Гарднера (с аномальным рубцеванием или без него).
Семейный аденоматозный полипоз может иметь разные модели наследования и разные генетические причины. Когда это состояние возникает в результате мутаций в гене APC, оно наследуется по аутосомно-доминантному типу , что означает, что одной копии измененного гена достаточно, чтобы вызвать нарушение. Заболеваемость злокачественными новообразованиями в этих случаях приближается к 100%. В большинстве случаев у больного есть один родитель с этим заболеванием.
APC представляет собой ген-супрессор опухоли, ответственный за продукцию аденоматозного полипоза coli (APC), большой многофункциональный белок, подавляющий опухоль, действует как «привратник», который предотвращает развитие опухолей. (APC регулирует β-β-поленин, белок, который играет решающую роль в клеточной коммуникации, передаче сигналов, росте и управляемом разрушении, но который, оставаясь неконтролируемым, также вызывает многочисленные раковые заболевания ). Недостаток в гене APC означает, что APC не так эффективен, как должен быть, и со временем вполне вероятно, что некоторые клетки, которые должны действовать, APC, перестанут действовать, а вместо этого будут продолжать развиваться и становиться злокачественными. При знакомом полипозе они обычно проявляются как полипы - небольшие аномалии на поверхности кишечного тракта.
Хотя полипы по своей природе доброкачественные, это первый шаг гипотезы двух ударов уже произошло: унаследованная мутация APC. Часто оставшийся «нормальный» аллель мутируется или удаляется, что ускоряет образование полипов. Дальнейшие мутации (например, в p53 или kRAS) в APC-мутантных клетках с большей вероятностью приведут к раку, чем в немутантных эпителиальных клетках.
Нормальное функционирование продукта гена APC все еще исследуется; он присутствует как в ядре клетки, так и в мембране. Каноническая опухолевая супрессорная функция APC заключается в подавлении β-катенина, но другие опухолевые супрессорные функции связаны с клеточной адгезией и организацией цитоскелета.
Мутация APC также часто встречается в отдельных случаях колоректальной карциномы, что подчеркивает ее важность при этой форме рака.
MUTYH кодирует фермент репарации ДНК. Во время нормальной клеточной активности гуанин иногда вызывает под действием кислорода, что заставляет его спариваться с аденином вместо цитозина. Гликозилаза MYH исправляет эти ошибки посредством эксцизионной репарации оснований, так что мутации не накапливаются в ДНК и не приводят к образованию опухоли. Когда гликозилаза MYH функционирует должным образом, возникла ошибка ДНК, чтобы инициировать онкогенез с клиническими проявлениями, аналогичными таковыми у пациентов с пациентами APC.
Мутации в гене MUTYH наследуются по аутосомно-рецессивному образцу, что означает, что две копии гена должны быть друг, чтобы человек пострадал от заболеваний. Чаще всего родители ребенка с аутосомно-рецессивным заболеванием не страдают, но являются носителями одной копии измененного гена.
Модель мыши «Apc» былаана в 1990 году и несет аллель Apc со стоп-кодоном в положении 850. Гетерозиготность этой мутации приводит к полностью пенетрантному фенотипу в большинстве генетических фонов, с мышами на чувствительном фоне, у которых развивается более 100 опухолей в кишечном тракте. Количество и расположение кишечника изменяются несвязанными генами. С тех пор появилось много других моделей, включая модель ослабленного FAP (модель 1638N) и несколько, которые позволяют тканеспецифичную или временную абляцию функции гена. Для получения дополнительной информации см. мышиные модели колоректального и кишечного рака.
В 2007 году модель крыс «ApcPirc» была выделена со стоп-кодоном в положении 1137. В отличие от мышиных моделей, где>90% опухолей формируются в тонком кишечнике крысы Pirc образуют опухоли преимущественно (>60%) в толстом кишечнике, аналогично клиническим проявлениям у человека.
 Микрофотография канальцевой аденомы, предшественника колоректального рака, который чаще всего ассоциируется с FAP
Микрофотография канальцевой аденомы, предшественника колоректального рака, который чаще всего ассоциируется с FAP Важно диагностировать FAP до развития рака толстой кишки не только для отдельного человека, но и для других членов семьи, которые могут пострадать. Существуют два диагностических метода:
NCBI заявляет, что врачи должны убедиться, что они понимают «риски, преимущества и ограничения» любого генетического теста, проведенного с 1997 года, «почти для одной трети людей, прошедших оценку на FAP, врач неверно истолковал результаты теста».
После постановки диагноза ФАП требуется тщательное колоноскопическое наблюдение с полипэктомией.
Пренатальное тестирование возможно, если у пораженного члена семьи выявлена мутация, вызывающая заболевание; тем не менее, пренатальное тестирование для лечения расстройств, начинающихся обычно у взрослых, встречается редко и требует тщательного генетического консультирования.
УЗИ брюшной полости и анализа крови для оценки функции печени часто. выполнено для исключения метастазов в печень.
 Колэктомия наличие образца, показывающего полипы по всей толстой кишке
Колэктомия наличие образца, показывающего полипы по всей толстой кишке Из-за того, как возможно семейный полипоз, генетические заболевания и, следовательно, риск, но никаких полипов или проблем пока нет. Следовательно, человеку может быть поставлен диагноз "риск" FAP, и ему требуется регулярное наблюдение, но он (пока) не имеет FAP (то есть есть дефектный ген, но, по-видимому, у него пока нет никаких реальных медицинских проблем в результате этого).). Клиническое ведение может охватывать несколько случаев:
NCBI заявляет, что «Хотя у многих людей с диагнозом полипоза, ассоциированного с APC, есть пораженный родитель, семейный анамнез может оказаться отрицательным из-за неспособности распознать расстройство у членов, ранняя смерть родителя» до появления симптомов или позднее начало заболевания у пораженного родителя ». Кроме того, около 20% случаев являются мутациями de novo, а из тех, у которых есть очевидная мутация APC de novo (т.е. неизвестный семейный анамнез), 20% имеют соматический мозаицизм. Известно также, что существуют бессимптомные люди (и, следовательно, бессимптомные члены семьи).
Мониторинг включает в себя амбулаторную колоноскопию, а иногда и верхних отделов желудочного тракта эзофагогастродуоденоскопия (EGD, для поиска предраковых опухолей желудка или двенадцатиперстной кишки ), обычно один раз в 1-3 года, и / или генетический анализ крови для окончательного подтверждения или опровержения предрасположенности. Небольшое количество полипов часто можно иссечь (удалить) во время процедуры, если они обнаружены, но при наличии более серьезных признаков или цифр может потребоваться стационарное хирургическое вмешательство.
NCBI заявляет, что, когда у человека идентифицирован FAP или мутации, приводящие к FAP: «Уместно оценить родителей пострадавшего человека (а) с помощью молекулярно-генетического тестирования APC, если заболевание - вызывающая мутация известна у пробанда [человека, впервые идентифицированного с заболеванием] или (б) для клинических проявлений состояний полипоза, ассоциированного с APC ».
Лечение для FAP зависит от генотипа. У большинства людей с мутацией APC рак толстой кишки развивается к 40 годам, хотя менее распространенная аттенуированная версия обычно проявляется в более позднем возрасте (40–70). Соответственно, во многих случаях профилактическое хирургическое вмешательство может быть рекомендовано до достижения 25-летнего возраста или после обнаружения при активном наблюдении. Существует несколько хирургических вариантов, которые включают удаление толстой или прямой и толстой кишки.
Профилактика колэктомия показан при наличии более ста полипов, при наличии полипов с тяжелой дисплазией или при наличии нескольких полипов размером более 1 см.
Лечение двух более легких формFAP может отличаться от более обычного варианта, так как количество полипов намного меньше, что дает больше возможностей.
В настоящее время исследуются лекарства для замедления качественного перерождения полипов, в первую очередь нестероидные противовоспалительные препараты (НПВП). Было показано, что НПВП значительно уменьшают количество полипов, но обычно не действует на лечение, поскольку еще много полипов, как это происходит и лечит эндоскопически.
До достижения поздних стадий колоректального рака полипы ограничены внутренней стенкой и толщиной кишечного тракта и не дают метастазов и не «распространяются». Таким образом, при условии, что FAP выявляется и контролируется либо на предраковой стадии, либо когда какие-либо раковые полипы находятся внутри кишечного тракта, операция имеет очень высокий уровень успеха в предотвращении или удалении рака без рецидива, поскольку места, вызывающие рак физически полностью удаляются хирургическим путем.
После операции, если была выполнена частичная колэктомия, необходимо колоноскопическое наблюдение за оставшейся ободочной кишкой, поскольку у человека все еще есть риск развития рака толстой кишки. Однако, если бы это произошло, это был бы новый случай полипов, вновь в не удаленной части толстой кишки после операции, а не возврат или метастаз любого рака, удаленной первоначальной операцией.
Распространенность мутации составляет от 1 на 10 000 до 1 на 15 000 рождений. К 35 годам у 95% людей с ФАП (>100 аденом) появляются полипы. Без колэктомии рак толстой кишки неизбежен. Средний возраст рака толстой кишки у нелеченных лиц составляет 39 лет (от 34 до 43 лет).
Ослабленный FAP, когда APC неисправен, но все еще работает. В результате он частично сохраняет способность подавлять полипы. Следовательно, ослабленный FAP проявляется как колоректальный рак необычно поздно (возраст 40–70, в среднем = 55) и, как правило, с небольшим или по крайней мере, гораздо меньшим полипов (обычно 30), чем более обычная версия FAP, в среднем возрасте, когда FAP больше не считается большой вероятностью или риском согласно обычной эпидемиологии FAP.
В этой таблице сравниваются различные подтипы FAP:
| Элемент | FAP | Аттенуированный FAP | MUTYH-ассоциированный FAP | ||
| Ген | APC | APC | MUTYH | ||
| Типичное проявление полипа | Сотни / тысячи | Менее 100 (0–470, тип. 30), иногда плоской, а не полиповидной морфологии и проксимальнее селезеночного изгиба. В исследовании с участием 120 человек 37% (N = 44) имели <10 polyps; 3 of these 44 had colorectal cancer]. Gastric fundic polyps and duodenal adenomas are also seen. Therefore, polyps and cancers may manifest in the upper portion of the colon or upper gastrointestinal tract rather than the usual locations | ? | ||
| Типичные основные диагностические критерии | (a) 100+ полипов и возраст до 40 лет, ИЛИ (b) полипы и FAP в относительной | Еще не решено. (а) отсутствие в семейном анамнезе 100+ полипов до 30 лет ПЛЮС ОДИН ИЗ 10–99 полипов / 100+ полипов в возрасте от 35 до 40 / колоректальный рак до 60 лет и родственников с множественными аденоматозными полипами, ИЛИ (b) Семейный анамнез От 10 до 99 аденом, диагностированных после 30 лет | ? | ||
| Возраст, в котором полипы проявляются | 7–36 (тип. 16), после чего быстро возрастает | ? | ? | ||
| риск колоректального рака (пенетрантность ) и возраст при отсутствии лечения | «неизбежно… практически 100%»: 7% к 21 году, 87% к 45 годам, 93% к 50 годам. Типичный возраст: 34–43 (средний показатель 39) | «Ниже.. менее известна.. по оценкам 70% к 80 годам». По данным Sovaria по состоянию на 1998 год, «средний возраст при постановке диагноза CRC составляет ~ 58 лет» | ? | ||
| Вариабельность | Меж- и внутрисемейная фенотипическая изменчивость является обычным явлением | См. FAP | ? | ||
| «полипы дна желудка и двенадцатиперстной болезни, остеомы, аномалии зубов, врожденная гипертрофия пигментного эпителия сетчатки (CHRPE), опухоли мягких тканей, десмоидные опухоли и связанные с ними раковые заболевания» | Как для FAP, но «CHRPE и десмоидные опухоли встречаются редко», а также добавляется рак щитовидной железы. | ? | |||
| Другие пожизненные риски | «Карцинома тонкой кишки [двенадцатиперстной кишки или периампуллы] 4–12% [дистальнее двенадцатиперстной кишки] Редко; Аденокарцинома поджелудочной железы ~ 1%; папиллярная карцинома щитовидной железы 1-2%; ЦНС [тип. Медуллобластома] <1%; Liver hepatoblastoma 1.6%; Bile ducts adenocarcinoma Low but increased; Stomach adenocarcinoma <1% in Western cultures." | ? | ? | ||
| Наследование | "наследуется по аутосомно-доминантному типу. Приблизительно у 75% -80% людей с полипозом, ассоциированным с APC, есть пораженный родитель. Потомство пораженного человека подвержено 50% риску унаследовать мутацию, вызывающую | То же, что и FAP | Другой - рецессивный (носителями должны быть 2 родителя) | ||
| Генетический обзор и генетическое обнаружение | «Полное секвенирование всех экзонов APC и границ интрон-экзон наиболее точным из клинических тестов. Большинство мутаций APC являются бессмысленными мутациями или мутациями сдвига считывания, которые вызывают преждевременное усечение белка APC. Вероятность обнаружения мутации APC сильно зависит от тяжести полипоза толстой кишки и от семейного анамнеза. ◦ Примерно 20% людей с очевидным диагнозом. мутация APC de novo. Маркеры, используемые для анализа сцепления в условиях APC-ассоциированного полипоза, очень информативны и очень связаны с локусом APC; таким образом, их можно использовать с более чем 98% в более чем 95% семейном полипозе, ассоциированном с APC. Тестирование сцепления невозможно для семьи с одним заболеванием, которое часто возникает, когда у человека есть мутация гена de novo и нет проблемного потомства. Если не обнаружено заболевание, вызывающее мутацию APC, молекулярно-генетическое тестирование MUTYH (см. Диагноз). " | " Ожидается, что менее 30% людей с ослабленными фенотипами будут иметь идентифицируемую мутацию APC "(см. Также подробности в разделе FAP) | ? | ||
| Генотип-фенотип [основное состояние] | Наиболее частая мутация APC находится в кодоне 1309 и приводит к большому количеству полипов в раннем возрасте (~ 20). Сообщается об обильном полипозе (в среднем 5000) с мутациями в кодонах 1250–1464. наиболее частичные и целые Делеции APC связаны со 100–2000 аденом. Типичный возраст начала: от кодона 168 до 1580 (исключая 1309) = 30 лет, 5 'кодона 168 и 3' кодона 1580 = 52 года. | Ослабленный FAP связан с мутациями ( типичный ли усекающий) в 5'-частях гена (кодоны 1–177), экзоне 9 и дистальном 3'-конце гена; интерстициальные делеции хромосомы 5q22, включающие APC; частичные и целые делеции гена; и соматический мозаицизм для мутаций APC, обычно Совария заявляет, что аттенуированный FAP «вызван мутац» и экзями в трех разных областях гена БТР - 5'-конце в области, охватывающую 4 и 5, экзон 9 и крайний 3'-конец. Фенотипическая экспрессия в этих трех группах родственных связей является переменной, но определенно более мягкой, чем при классическом FAP "и что полипы прямой кишки редки при ослабленном FAP, но еще не подтверждено, означает ли это также, что риск рака прямой кишки также ниже. 262>Генотип - фенотип [Другие экстра-толстые состояния] | Выраженные экстраколонические проявления часто коррелируют (хотя и не полностью) более дистальными мутациями APC. Общее исследование FAP плюс экстраколонические симптомы показало : мутации в кодонах 1395–1493 используются более высокие частоту возникновения десмоидных опухолей, остеом и эпидермоидных кист, чем мутации с мутациями в кодонах 177–452; мутации в кодонах 1395–1493 имеют значительно более частые причины возникновения десмоидных опухолей и остеомов, чем мутации с мутациями в кодонах 177–452; в кодонах 457–1309; ни у одного человека с мутациями в кодонах 177–452 не развились остеомы или периампулярный рак; только у людей с мутациями в кодонах 457–1309 развиваются гепатобластома и опухоли головного мозга. Аденомы двенадцатиперстной кишки : четырехкратное повышение риска при мутациях между кодонами 976 и 1067. Десмоидные опухоли : мутации 3 ’в кодоне 1399 были связаны с развитием десмоидной опухоли с отношением шансов 4,37; десмоидные опухоли у 20% индивидов с мутациями 5 'в кодоне 1444, 49% индивидов с мутациями 3' в кодоне 1444 и 61% индивидов с мутациями в кодонах 1445–1580; несколько семей с тяжелыми десмоидными опухолями имели мутации на крайнем 3'-конце; последовательная ассоциация десмоидных опухолей с мутациями, дистальными по отношению к кодону 1444. CHRPE связан с: мутациями между кодонами 311 и 1444; целые делеции гена APC. Рак щитовидной железы и FAP : У 24 человек типичных идентифицированных мутаций относились к кодону 1220 от 5 '[Cetta et al. 2000]; 9 из 12 человек имели мутации APC, идентифицированные проксимальнее области кластера мутаций (кодоны 1286–1513). Общий обзор литературы (до августа 2006 г.) : выявлено 89 субмикроскопических делеций АРС (42 частных и 47 целых делеций гена). Экстраколонические находки наблюдались в 36% случаев, без существенных различий между частичными и целыми случаями гена. | ? | ? |
| Распространенность | "2,29–3,2 на 100 000 человек. Исторически сложилось так, что полипоз, связанный с APC, приходилось около 0,5% всех колоректального рака; эта цифра снижается, поскольку все " | " Вероятно, диагноз недооценен, учитывая меньшее количество полипов толстой кишки, и более низкий риск развития колоректального рака по сравнению с классическим FAP » | ? | ||
| Лечение проявлений | Классический FAP: «Колэктомия показывает после появления аденом; колэктомия может бытьена в зависимости от размера и количества аденоматозных полипов.» Колэктомия - это обычно рекомендуется, когда развивается более 20 или 30 аденом или множественных аденом « | » Может потребоваться колэктомия, но примерно для одной трети людей полипы толстой кишки достаточно ограничены в n отметьте, что наблюдение с периодической колоноскопиче ской полипэктомией достаточным " | ? | ||
| Наблюдение (мониторинг) после риска | " Ригмоидоскопия или колоноскопия каждые 1-2 года, начиная с возраста 10-12 лет; колоноскопия при обнаружении полипов; ежегодная колоноскопия, если колэктомия откладывается более чем на год после появления полипов (в возрасте от 10 до 20 лет с некоторыми более легкими симптомами, можно рассмотреть отсрочку колэктомии); Эзофагогастродуоденоскопия (ЭГДС) до 25 лет или до колэктомии и повторяется каждые 1–3 года; в некоторых случаях - эндоскопическая ретроградная холангиопанкреатография (ЭРХПГ) для оценки аденом общего желчного протока; визуализация тонкой кишки при обнаружении аденомы двенадцатиперстной кишки или перед колэктомией, повторение каждые 1–3 года в зависимости от результатов; скрининг на гепатобластому (другой интервал неизвестен, одна статья рекомендует «не реже одного раза в три месяца»); ежегодный медицинский осмотр, включая оценку внекишечных проявлений и пальпацию щитовидной железы с учетом последующего ультразвукового исследования и тонкоигольной аспирации, если присутствуют узелки щитовидной железы " | " Колоноскопия каждые два-три года, начиная с возраста от 18 до 20 лет; эзофагогастродуоденоскопия (ЭГДС), начинающаяся в возрасте 25 лет или до колэктомии и повторяющаяся каждые 1–3 года; в некоторых случаях может потребоваться эндоскопическая ретроградная холангиопанкреатография (ERCP); ежегодный медицинский осмотр с пальпацией щитовидной железы с учетом повторного ультразвукового исследования и тонкоигольной аспирации при наличии узлов щитовидной железы. Колэктомия рекомендуется при развитии более 20 или 30 аденом или множественных аденом с развитой гистологией ». Sovaria по состоянию на 1998 год заявляет, что «колоноскопия, в отличие от ректороманоскопии, должна быть рекомендована для эндоскопического наблюдения из-за правостороннего расположения колоректального отдела». аденомы; Эндоскопическое наблюдение УГИ оправдано в попытке попытки предраковые опухоли желудоли или двенадцатиперстной кишки; лицам, страдающим [ослабленным ФАП], может потребоваться полная колэктомия с илео-ректальным анастомозом только в том случае, если рекомендована профилактическая колэктомия » | ? | ||
| Решение о мониторинге | « Раннее распознавание может позволить своевременное вмешательство и улучшить конечный результат; таким образом, наблюдение за бессимптомными детьми группы риска на предмет ранних проявлений является целесообразным; генетическое тестирование более рентабельно, чем ректороманоскопия, для определения того, кто из членов семьи пострадал; Лица, у которых диагностирован полипоз, связанный с APC, в результате наличия у пострадавшего родственника, имеют значительно большую ожидаемую продолжительность жизни, чем люди, у которых диагностирован диагноз на основе симптомов. Поскольку мониторинг толстой кишки для лиц с риском классического FAP начинается уже в возрасте от десяти до 12 лет, молекулярно-генетическое тестирование обычно предлагается детям с риском классического ФАП к возрасту 10 лет. Также может потребоваться генетическое тестирование при рождении, поскольку некоторые родители и педиатры могут рассмотреть возможность проведения скрининга на гепатобластому с младенчества до пятилетнего возраста у пораженного потомства. Нет доказательств, указывающих на оптимальный возраст для начала скрининга. " | См. FAP. Также «Скрининг толстой кишки для пациентов с ослабленным FAP начинается в возрасте от 18 до 20 лет; таким образом, молекулярно-генетическое тестирование должно быть предложено тем, кто подвержен риску аттенуированного FAP примерно в возрасте 18 лет ». | ? | ||
| Наследование и последствия подтвержденного диагноза для других близких родственников | APC-ассоциированный полипоз наследуется в аутосомно-доминантный характер. Примерно 20–25% имеют измененный ген в результате мутации гена de novo. Отсутствие или отсутствие доказательств предвзятости по материнскому / отцовскому признаку или эффекта, связанного с преклонным отцовским возрастом, при мутациях de novo. % -ный риск совместного заболевания, если он унаследован, а не de novo и «низкий», но немного более высокий риск, чем общий, если de novo, поэтому следует предложить генетическое тестирование. У каждого потомства есть 50% -ный шанс наследования. Другие члены семьи находятся в группе риска если у их родителей одна и та же мутация. Мозаицизм зародышевой линии был зарегистрирован в бессимптомных случаях. Пренатальное тестирование возможно с помощью извлеченной из плода ДНК. | См. FAP | ? |
Из-за генетического характер ФАП, реестры полипозов разработаны по всему миру. Целью этих реестров является повышение осведомленности о передаваемости FAP, а также отслеживание семейных поражений. Одно исследование показало, что использовать для уведомления пользователей (звонков) снижает смертность по сравнению с пробандами. Св. Регистр полипоза Марка является старейшим в мире, он был запущен в 1924 году, и в настоящее время существует множество других регистров полипоза.
| Классификация | D |
|---|---|
| Внешние ресурсы |