
Войти
| Синдром Хантера | |
|---|---|
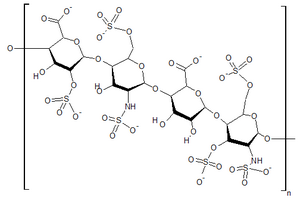 | |
| Структура гепарансульфата, одного из ГАГ, который накапливается в тканях людей с синдромом Хантера. | |
| Специальность | Эндокринология |
| Симптомы | Скелетные аномалии, потеря слуха, дегенерация сетчатки, увеличение печени и селезенки |
| Осложнения | Заболевания верхних дыхательных путей; сердечно-сосудистая недостаточность |
| Причины | Дефицит фермента идуронат-2-сульфатазы |
| Дифференциальная диагностика | Мукополисахаридоз I типа ; другие мукополисахаридозы |
| Прогноз | В тяжелых случаях смерть обычно наступает к 15 годам. В более тяжелых случаях пациенты могут дожить до 50 лет. |
| Частота | 1 из 100 000–150 000 новорожденных мальчиков |
Синдром Хантера, или мукополисахаридоз типа II ( MPS II), является редким генетическим заболеванием, при котором в тканях организма накапливаются большие молекулы сахара, называемые гликозаминогликанами (или ГАГ или мукополисахариды). Это форма лизосомальной болезни накопления. Синдром Хантера вызван дефицитом лизосомального фермента идуронат-2-сульфатазы (I2S). Недостаток этого фермента заставляет гепарансульфат и дерматансульфат накапливаться во всех тканях организма. Синдром Хантера является единственным синдромом МПС с X-сцепленным рецессивным наследованием.
Симптомы синдрома Хантера сравнимы с теми из МПС I. Это вызывает аномалии во многих органах, включая скелет, сердце и дыхательную систему. В тяжелых случаях это приводит к летальному исходу в подростковом возрасте. В отличие от MPS I, помутнение роговицы не связано с этим заболеванием.
Синдром Хантера может иметь широкий спектр фенотипов. Он традиционно классифицируется как «легкий» или «тяжелый» в зависимости от наличия симптомов со стороны центральной нервной системы, но это чрезмерное упрощение. Пациенты с «ослабленной» или «легкой» формой заболевания могут по-прежнему страдать от серьезных проблем со здоровьем. Для пациентов с тяжелым поражением клиническое течение относительно предсказуемо; пациенты обычно умирают в раннем возрасте. Для людей с более легкими формами болезни существует более широкий спектр исходов. Многие доживают до 20-30 лет, но у некоторых средняя продолжительность жизни может быть почти нормальной. Сердечные и респираторные нарушения - обычная причина смерти пациентов с более легкими формами заболевания.
Симптомы синдрома Хантера (МПС II) обычно не проявляются при рождении. Часто первые симптомы могут включать грыжи живота, ушные инфекции, насморк и простуду. Поскольку накопление ГАГ продолжается во всех клетках тела, признаки MPS II становятся более заметными. Внешний вид многих детей с синдромом включает характерную грубость черт лица, в том числе выступающий лоб, нос со сплющенной переносицей и увеличенный язык. У них также может быть большая голова, а также увеличенный живот. В тяжелых случаях МПС II диагноз часто ставится в возрасте от 18 до 36 месяцев. В более легких случаях пациенты проявляют себя так же, как дети с синдромом Херлера – Шейе, и диагноз обычно ставится в возрасте от 4 до 8 лет.
Продолжительное хранение ГАГ приводит к нарушениям во многих системах органов. После 18 месяцев дети с тяжелым МПС II могут страдать от снижения развития и прогрессирующей потери навыков. Утолщение сердечных клапанов и стенок сердца может привести к прогрессирующему ухудшению сердечной функции. Стенки дыхательных путей также могут утолщаться, что приводит к обструктивному заболеванию дыхательных путей. Поскольку печень и селезенка со временем увеличиваются в размерах, живот может увеличиваться в размерах, что делает грыжи более заметными. MPS II может затронуть все основные суставы, что приведет к их скованности и ограничению движений. Прогрессирующее поражение суставов пальцев и большого пальца приводит к снижению способности подбирать мелкие предметы. Воздействие на другие суставы, такие как бедра и колени, может затруднить нормальную ходьбу. Если развивается синдром запястного канала, может произойти дальнейшее снижение функции руки. Сами кости могут быть поражены, что приведет к низкому росту. Кроме того, у некоторых людей с ним могут быть обнаружены галечные поражения кожи цвета слоновой кости на предплечьях, ногах и верхней части спины. Эти поражения кожи считаются патогномоничными для заболевания. Наконец, накопление ГАГ в головном мозге может привести к задержке развития с последующей умственной отсталостью и прогрессирующей потерей функции.
Возраст появления симптомов и наличие или отсутствие поведенческих нарушений являются прогностическими факторами окончательной тяжести заболевания у очень маленьких пациентов. Нарушения поведения часто могут имитировать комбинации симптомов синдрома дефицита внимания с гиперактивностью, аутизма, обсессивно-компульсивного расстройства и / или расстройства обработки сенсорной информации, хотя наличие и уровень симптомов различаются у каждого пораженного ребенка. Они также часто включают отсутствие надлежащего чувства опасности и агрессии. Поведенческие симптомы MPS II обычно предшествуют нейродегенерации и часто усиливаются до тех пор, пока психические расстройства не становятся более выраженными. К моменту смерти большинство детей с тяжелым МПС II имеют тяжелые психические расстройства и полностью зависят от своих опекунов.
 MPS II имеет рецессивный паттерн наследования, сцепленный с Х-хромосомой.
MPS II имеет рецессивный паттерн наследования, сцепленный с Х-хромосомой. Поскольку синдром Хантера является Х-сцепленным рецессивным заболеванием, он преимущественно поражает пациентов мужского пола. Ген IDS расположен на Х-хромосоме. Ген IDS кодирует фермент идуронат-2-сульфатаза (I2S). Недостаток этого фермента приводит к накоплению ГАГ, которые вызывают симптомы MPS II. У женщин обычно две Х-хромосомы, тогда как у мужчин обычно одна Х-хромосома, которую они наследуют от своей матери, и одну Y-хромосому, которую они наследуют от своего отца.
Если самка наследует одну копию мутантного аллеля MPS II, у нее обычно будет нормальная копия гена IDS, которая может компенсировать мутантный аллель. Это известно как генетический носитель. Однако мужчина, унаследовавший дефектную Х-хромосому, обычно не имеет другой Х-хромосомы, компенсирующей мутантный ген. Таким образом, женщина должна унаследовать два мутантных гена для развития MPS II, в то время как пациенту мужского пола необходимо унаследовать только один мутантный ген. Женщина-носитель может пострадать из-за X-инактивации, которая является случайным процессом.
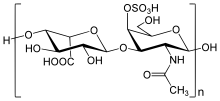 Дерматансульфат - один из ГАГ, который накапливается в тканях людей с МПС II.
Дерматансульфат - один из ГАГ, который накапливается в тканях людей с МПС II. Человеческое тело зависит от огромного множества биохимических реакций для поддержки критически важных функций. Одна из этих функций - разрушение больших биомолекул. Неудача этого процесса - основная проблема синдрома Хантера и связанных с ним нарушений памяти.
Биохимия синдрома Хантера связана с проблемой в части соединительной ткани, известной как внеклеточный матрикс, которая состоит из различных сахаров и белков. Помогает сформировать архитектурный каркас тела. Матрица окружает клетки тела организованной сеткой и действует как клей, скрепляющий клетки тела. Одна из частей внеклеточного матрикса - это молекула, называемая протеогликаном. Как и многие компоненты организма, протеогликаны необходимо расщеплять и заменять. Когда организм расщепляет протеогликаны, одним из образующихся продуктов являются мукополисахариды (ГАГ).
В MPS II проблема касается разрушения двух ГАГ: дерматансульфата и гепарансульфата. На первом этапе расщепления дерматансульфата и гепарансульфата необходим лизосомальный фермент идуронат-2-сульфатаза, или I2S. У людей с МПС II этот фермент частично или полностью неактивен. В результате ГАГ накапливаются в клетках по всему телу, особенно в тканях, которые содержат большое количество дерматансульфата и гепарансульфата. Скорость накопления ГАГ не одинакова для всех людей с MPS II, что приводит к широкому спектру медицинских проблем.
Первый лабораторный скрининговый тест на расстройство MPS - это анализ мочи на ГАГ. Аномальные значения указывают на вероятность нарушения МПС. Иногда анализ мочи может быть нормальным, даже если у ребенка действительно есть расстройство МПС. Окончательный диагноз MPS II ставится путем измерения активности I2S в сыворотке, лейкоцитах или фибробластах при биопсии кожи. У некоторых людей с MPS II анализ гена I2S может определить клиническую тяжесть.
Пренатальная диагностика обычно доступна путем измерения ферментативной активности I2S в околоплодных водах или ткани ворсинок хориона. Если известно, что в семье присутствует конкретная мутация, можно провести пренатальное молекулярно-генетическое тестирование. Секвенирование ДНК может выявить, является ли кто-то носителем болезни.
Из-за большого разнообразия фенотипов лечение этого расстройства определяется специально для каждого пациента. До недавнего времени не существовало эффективной терапии MPS II, поэтому использовалась паллиативная помощь. Однако последние достижения привели к созданию лекарств, которые могут улучшить выживаемость и благополучие людей с МПС II.
Идурсульфаза, очищенная форма недостающего лизосомального фермента, прошла клинические испытания в 2006 году и впоследствии была одобрена Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США в качестве ферментативной терапии для лечения МПС II. Идурсульфаза бета, еще одно заместительное лечение фермента, было одобрено в Корее Министерством продовольствия и безопасности лекарств.
Доказано, что недавние достижения в заместительной ферментной терапии ( ФЗТ ) с идурсульфазой улучшают многие признаки и симптомы МПС II, особенно если ее начать на ранней стадии заболевания. После введения его можно транспортировать в клетки для расщепления ГАГ, но, поскольку лекарство не может преодолевать гематоэнцефалический барьер, не ожидается, что оно приведет к улучшению когнитивных функций у пациентов с тяжелыми симптомами со стороны центральной нервной системы. Даже с помощью ФЗТ необходимо лечение различных проблем органов у самых разных медицинских специалистов.
Трансплантация костного мозга и трансплантация гемопоэтических стволовых клеток (HSCT) использовались в качестве лечения в некоторых исследованиях. Хотя трансплантация принесла пользу многим системам органов, не было показано, что она улучшает неврологические симптомы заболевания. Хотя ТГСК показала себя многообещающим при лечении других расстройств МПС, его результаты до сих пор были неудовлетворительными при лечении МПС II. Было показано, что ФЗТ приводит к лучшим результатам у пациентов с МПС II.
В феврале 2019 года ученые-медики, работающие с Sangamo Therapeutics, штаб-квартира которой находится в Ричмонде, штат Калифорния, объявили о первой терапии по редактированию генов человека для постоянного изменения ДНК у пациента с МПС II. Клинические испытания Sangamo, включающие редактирование генов с использованием нуклеазы цинковых пальцев, продолжаются по состоянию на февраль 2019 года.
Более раннее появление симптомов связано с худшим прогнозом. У детей, у которых проявляются симптомы в возрасте от 2 до 4 лет, смерть обычно наступает в возрасте от 15 до 20 лет. Причина смерти обычно связана с неврологическими осложнениями, обструктивным заболеванием дыхательных путей и сердечной недостаточностью. Если пациенты имеют минимальные неврологические нарушения, они могут дожить до 50 лет и старше.
По оценкам, 2000 человек во всем мире страдают MPS II, 500 из которых живут в Соединенных Штатах.
Исследование, проведенное в Соединенном Королевстве, показало, что заболеваемость среди мужчин составляет около одного из 130 000 живорождений мужского пола.
Синдром назван в честь врача Чарльза А. Хантера (1873–1955), который впервые описал его в 1917 году.
Начиная с 2010 года, в рамках фазы I / II клинических испытаний оценивали интратекальные инъекции более концентрированной дозы идурсульфазы, чем внутривенный состав, используемый в инфузиях заместительной ферментной терапии, в надежде предотвратить снижение когнитивных функций, связанное с тяжелой формой состояния. Результаты были представлены в октябре 2013 года. В 2014 году началась фаза II / III клинических испытаний.
В 2017 году 44-летний пациент с МПС II прошел курс генной терапии в попытке предотвратить дальнейшее повреждение от болезни. Это первый случай использования генной терапии у людей in vivo. В 2018 году исследование было распространено на шесть пациентов.
С 24 июля 2004 года, Эндрю Wragg, 38, из Worthing, Западный Суссекс, Англия, задушили его 10-летний сын Иакова с подушкой, из - за инвалидности мальчика, связанные с МПС II. Военная безопасность специалист, Wragg также утверждал, что он был в состоянии стресса после возвращения с войны в Ираке. Он отрицал убийство Иакова, но признал себя виновным в непредумышленном убийстве из-за ограниченной дееспособности. Миссис судья Энн Рафферти назвала случай «исключительным», приговорила Ррагга к двум годам тюремного заключения за непредумышленное убийство, а затем условно на два года приговорила его. Рафферти сказал, что «ничего не выиграет» от отправки Рэгга в тюрьму за преступление.
| Классификация | D |
|---|---|
| Внешние ресурсы |