
Войти
| Болезнь Урбаха-Вите | |
|---|---|
| Другие имена | Липоидный протеиноз и гиалиноз кожи и слизистых оболочек |
 | |
| Болезнь Урбаха – Вите наследуется по аутосомно-рецессивному типу. | |
| Специальность | Эндокринология |
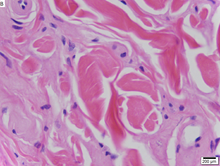 Болезнь Урбаха – Вите при биопсии кожи с окрашиванием Hamp;E.
Болезнь Урбаха – Вите при биопсии кожи с окрашиванием Hamp;E. Болезнь Урбаха-Вите - очень редкое рецессивное генетическое заболевание, с момента его обнаружения зарегистрировано около 400 случаев. Впервые об этом официально сообщили в 1929 году Эрих Урбах и Камилло Вите, хотя случаи, возможно, относятся к 1908 году.
Симптомы болезни сильно различаются от человека к человеку. Они могут включать хриплый голос, повреждения и рубцы на коже, легко повреждаемую кожу с плохим заживлением ран, сухую, морщинистую кожу и образование бусинок на папулах вокруг век. Все это результат общего утолщения кожи и слизистых оболочек. В некоторых случаях также наблюдается отвердение мозговой ткани в медиальных височных долях, что может привести к эпилепсии и психоневрологическим нарушениям. Заболевание, как правило, не опасно для жизни, и у пациентов не наблюдается снижения продолжительности жизни.
Поскольку болезнь Урбаха – Вите является аутосомно-рецессивным заболеванием, люди могут быть носителями болезни, но не проявлять никаких симптомов. Заболевание вызвано мутациями потери функции в хромосоме 1 в гене 1q21, гене белка внеклеточного матрикса 1 ( ECM1 ). Дерматологические симптомы вызваны накоплением гиалинового материала в дерме и утолщением базальных мембран кожи. Болезнь Урбаха-Вите обычно диагностируется по ее клиническим дерматологическим проявлениям, особенно по бусинчатым папулам на веках. Обнаружение мутаций в гене ECM1 позволило использовать генетическое тестирование для подтверждения первоначального клинического диагноза. Для диагностики также можно использовать периодическую кислоту-Шиффа (PAS) и иммуногистохимическое окрашивание.
В настоящее время не существует лекарства от болезни Урбаха – Вите, хотя есть способы индивидуального лечения многих ее симптомов. Открытие мутаций гена ECM1 открыло возможность генной терапии или рекомбинантного белка ECM1 для лечения болезни Урбаха-Вите, но ни один из этих вариантов в настоящее время недоступен. Некоторые исследователи обследуют пациентов с болезнью Урбаха – Вите, чтобы узнать больше о других состояниях, которые проявляют похожие неврологические симптомы, например аутизм.
Болезнь Урбаха – Вите характеризуется как неврологическими, так и дерматологическими симптомами.
Хотя симптомы могут сильно различаться у разных людей, даже у членов одной семьи, симптомы обычно начинаются в младенчестве и обычно являются результатом утолщения кожи и слизистых оболочек. Первым симптомом часто бывает слабый крик или хриплый голос из-за утолщения голосовых связок. Хриплый голос может быть один из самых ярких клинических проявлений заболевания. Поражения и рубцы также появляются на коже, обычно на лице и дистальных отделах конечностей. Это часто является результатом плохого заживления ран, и с возрастом рубцы продолжают увеличиваться, оставляя кожу воскообразной. Кожу можно легко повредить в результате незначительной травмы или травмы, оставив множество волдырей и дополнительных шрамов. Кожа также обычно бывает очень сухой и морщинистой. Белые или желтые инфильтраты образуются на губах, слизистой оболочке щек, миндалинах, язычке, надгортаннике и уздечке языка. Это может привести к инфекции верхних дыхательных путей и иногда требует трахеостомии для облегчения симптома. Слишком сильное утолщение уздечки может ограничить движение языка и привести к затруднениям речи. Вальца, папулы вокруг века является очень распространенным симптомом и часто используются как часть диагностики этого заболевания. Некоторые другие дерматологические симптомы, которые иногда наблюдаются, но встречаются реже, включают выпадение волос, паротит и другие стоматологические аномалии, язвы роговицы и очаговую дегенерацию желтого пятна.
Хотя дерматологические изменения являются наиболее очевидными симптомами болезни Урбаха – Вите, у многих пациентов также наблюдаются неврологические симптомы. Около 50–75% диагностированных случаев болезни Урбаха – Вите также показывают двусторонние симметричные кальцификаты на медиальных височных долях. Эти обызвествления часто влияют на миндалину и periamygdaloid извилины. Считается, что миндалевидное тело участвует в обработке биологически значимых стимулов и в долговременной эмоциональной памяти, особенно связанных со страхом, и ПЭТ- и МРТ- сканирование показали корреляцию между активацией миндалины и эпизодической памятью на сильные эмоциональные стимулы. Следовательно, пациенты с болезнью Урбаха-Вите с кальцификациями и поражениями в этих регионах могут страдать нарушениями в этих системах. Эти кальцификации являются результатом накопления отложений кальция в кровеносных сосудах этой области мозга. Со временем эти сосуды затвердевают, и ткань, в которую они входят, отмирает, вызывая поражения. Количество кальциноза часто зависит от продолжительности заболевания. Трудно точно определить истинную распространенность этих кальцификатов, поскольку не всем пациентам проводится томография головного мозга. У некоторых пациентов также наблюдаются эпилепсия и психоневрологические нарушения. Симптомы эпилепсии могут начаться с приступов легкого беспокойства, и их можно контролировать с помощью противоэпилептических препаратов. Другие пациенты имеют симптомы, похожие на шизофрению, в то время как некоторые страдают настроением, тревогой и психотическими расстройствами.
 Хромосома 1, где мутация ECM1 происходит через 1 q 21
Хромосома 1, где мутация ECM1 происходит через 1 q 21 Исследователи сопоставили болезнь Урбаха-Вите с хромосомой 1 в точке 1q21 и конкретно идентифицировали ген белка внеклеточного матрикса 1 ( ECM1 ) как ген, содержащий мутации, которые могут привести к развитию этого состояния. На данный момент 41 различная мутация внутри ECM1, как сообщается, приводит к болезни Урбаха-Вите. Все они были гомозиготными с потерей функции мутации (то есть нонсенс, со сдвигом рамки или внутренние делеции ). Это аутосомно-рецессивное заболевание, требующее наличия двух мутировавших копий гена ECM1, чтобы вызвать заболевание.
ECM1 кодирует гликопротеин ранее неизвестного происхождения. Открытие того, что потеря экспрессии ECM1 приводит к симптомам, связанным с болезнью Урбаха-Вите, предполагает, что ECM1 может вносить вклад в адгезию кожи, дифференцировку эпидермиса и заживление ран и рубцевание. Также считается, что он играет роль в формировании эндохондральной кости, биологии опухолей, пролиферации эндотелиальных клеток и формировании кровеносных сосудов.
Дерматологические симптомы вызваны накоплением гиалинового материала в дерме и утолщением базальных мембран кожи. Природа этого материала неизвестна, но исследователи предположили, что это может быть гликопротеин, гликолипид, кислый мукополисахарид, измененный коллаген или эластичная ткань.
Болезнь Урбаха-Вите обычно диагностируется по ее клиническим дерматологическим проявлениям, в частности по бусинчатым папулам на веках. Врачи также могут протестировать гиалиновый материал с помощью периодического окрашивания кислотой Шиффа (PAS), так как материал сильно окрашивается для этого пятна.
Иммуногистохимическая маркировка кожи антител к белку ECM1, как было показано, снижается в коже людей, пораженных болезнью Урбаха-Вите. Окрашивание антителами к коллагену IV типа или антителами к коллагену VII типа выявляет яркие толстые полосы на дермоэпидермальном соединении.
Неконтрастная компьютерная томография может отображать кальцификаты, но обычно это не используется в качестве средства диагностики заболевания. Частично это связано с тем, что не у всех пациентов Урбаха-Вите наблюдаются кальцификаты, а также потому, что подобные поражения могут быть образованы при других заболеваниях, таких как простой герпес и энцефалит. Обнаружение мутаций в гене ECM1 позволило использовать генетическое тестирование для подтверждения первоначального клинического диагноза болезни Урбаха – Вите. Это также позволяет врачам лучше различать болезнь Урбаха – Вите и другие подобные заболевания, не вызванные мутациями в ECM1.
В настоящее время не существует лекарства от болезни Урбаха – Вите, хотя есть несколько способов индивидуального лечения многих ее симптомов. Был достигнут некоторый успех при пероральном приеме диметилсульфоксида (ДМСО) и гепарина внутри очага поражения, но это верно не во всех случаях. Д- пеницилламин также показал себя многообещающим, но пока не получил широкого применения. Также есть сообщения о пациентах, которых лечили этретинатом, лекарством, которое обычно назначают для лечения псориаза. В некоторых случаях кальцификаты в головном мозге могут приводить к аномальной электрической активности нейронов. Некоторым пациентам назначают противосудорожные препараты, чтобы помочь справиться с этими отклонениями. Трахеостомия часто используется для облегчения инфекций верхних дыхательных путей. Операция с использованием углекислотного лазера утолщенных голосовых связок и папул век, украшенных бусинами, улучшила эти симптомы у пациентов. Открытие мутаций гена ECM1 открыло возможность генной терапии или рекомбинантного белка EMC1 для лечения болезни Урбаха-Вите, но ни один из этих двух вариантов в настоящее время недоступен.
Болезнь Урбаха-Вите обычно не представляет угрозы для жизни. Ожидаемая продолжительность жизни этих пациентов является нормальной, если должным образом устранены потенциальные побочные эффекты утолщения слизистой оболочки, такие как обструкция дыхательных путей. Хотя для этого может потребоваться трахеостомия или операция с использованием лазера на углекислом газе, такие шаги могут помочь гарантировать, что люди с болезнью Урбаха-Вите смогут жить полноценной жизнью. Было показано, что пероральный диметилсульфоксид (ДМСО) уменьшает повреждения кожи, помогая минимизировать дискомфорт для этих людей.
Болезнь Урбаха – Вите встречается очень редко; в медицинской литературе зарегистрировано менее 300 случаев. Хотя болезнь Урбаха-Вите встречается во всем мире, почти четверть зарегистрированных диагнозов приходится на Южную Африку. Многие из них есть у пациентов голландского, немецкого и койсанского происхождения. Считается, что эта высокая частота связана с эффектом основателя. Из-за рецессивной генетической причины и способности переносить болезнь без симптомов болезнь Урбаха-Вите часто передается по наследству. В некоторых регионах Южной Африки до одного из 12 человек может быть носителем болезни. Большинство тематических исследований с участием пациентов с болезнью Урбаха – Вите включают от одного до трех случаев, и эти случаи часто происходят в одной семье. Из-за низкой заболеваемости трудно найти достаточно большое количество случаев для адекватного изучения болезни.
В 1908 году, что, как представляется, первый случай заболевания Урбах-Wiethe сообщил Friedrich Зибенмана, профессор отоларингологии в Базеле, Швейцария. В 1925 году швейцарский дерматолог Фридрих Мишер сообщил о трех похожих пациентах. Официальный отчет о болезни Урбаха-Вите был впервые описан в 1929 году венским дерматологом и оториноларингологом Урбахом и Вите. Его первоначальное название «lipoidosis cutis et mucosae» было изменено на «липоидный протеиноз cutis et mucosae» из-за убеждения Урбаха, что это состояние вызвано аномальными отложениями липидов и белков в тканях. Некоторые обсуждают, действительно ли это заболевание является формой мукополисахаридоза, амилоидоза или даже порфирии. Открытие болезни Урбаха-Вите, вызывающей мутацию гена ECM1, теперь предоставило окончательный способ дифференцировать болезнь Урбаха-Вите от этих других состояний.
| Классификация | D |
|---|---|
| Внешние ресурсы |