
Войти
В органической химии кинетическое разрешение является средством различения двух энантиомеры в рацемической смеси. При кинетическом разрешении два энантиомера реагируют с разными скоростями реакции в химических реакций с хиральным катализатором или реагентом, в результате чего получается энантиообогащенный образец менее реакционноспособного энантиомера.. Отличие от хирального разрешения, кинетическое разрешение не зависит от различных свойств диастереомерных продуктов, а, скорее, от химических свойств рацемических исходных материалов. Этот энантиомерный избыток (э.и.) непрореагировавшего материала непрерывно увеличивается по мере образования большего количества продукта, достигая 100% непосредственно перед полным завершением реакции. Кинетическое разрешение зависит от различных реакционных способностей энантиомеров или энантиомерных комплексов. Кинетическое разрешение - это понятие в органической химии, установка хиральных молекул в органический синтезе. Реакции кинетического разделения с использованием синтетических реагентов и катализаторов гораздо менее распространены, чем использование ферментативного разрешения в применении к органическому синтезу, хотя за последние 30 лет был разработан ряд полезных синтетических методов.
Получено первое известное кинетическое разрешение Луи Пастер. После реакции водного рацемического аммония тартрата с плесенью из Penicillium glaucum он повторно изолировал оставшийся тартрат и обнаружил, что он левовращающий. Хиральные микроорганизмы, присутствующие в плесени, избирательно катализируют метаболизм (R, R) -тартрата, оставляя избыток (S, S) -тартрата.
Кинетическое разрешение синтетическим впервые было обнаружено Марквальдом и МакКензи в 1899 году при этерификации рацемического миндальная кислота с оптически активным (-) - ментолом. При избытке рацемической кислоты они наблюдали, что образование сложного эфира, полученного из (+) - миндальной кислоты, происходит быстрее, чем образование сложного эфира из (-) - миндальной кислоты. Непрореагировавшая кислота имеет небольшой избыток (-) - миндальной кислоты, и позднее было показано, что сложный эфир дает (+) - миндальную кислоту при омылении. Важность этого наблюдения заключалась в том, что теоретически, если бы использовалась половина эквивалента (-) - ментола, можно было бы приготовить высокоэнантиообогащенный образец (-) - миндальной кислоты. Это наблюдение достигается к успешному кинетическому разделу других хиральных кислот, началу использования кинетического разрешения в органической химии.

Кинетическое разрешение является возможным методом необратимой дифференциации пары энантиомеров за счет (первого) разные энергии активации. Хотя оба энантиомера находятся на одном и том же уровне свободной энергии Гиббса по определению, и продукты реакции с обоими энантиомерами также находятся на равных уровнях, 

Идеальное кинетическое разрешение - это такое разрешение, при котором реагирует только один энантиомер, то есть k R>>k S. селективность (и) кинетического разрешения соответствует с константами скорости реакции энантиомеров R и S, k R и k S соответственно, на s = к R/kSдля к R>kS. Эту селективность также можно обозначать как относительные скорости реакции . Это можно записать в терминах разности свободной энергии между состояниями переходов с высокой и низкой энергией: 

Селективность также восстановленного исходного материала и конверсии (c), если кинетика первого порядка (в субстрате). Если судом, что энантиомер S исходного материала рацемат будет извлечен в избытке, можно выразить (мольные доли) S и R как
![[S] = {\ frac {(1 + ee) (1-c)} {2 }}](https://wikimedia.org/api/rest_v1/media/math/render/svg/bedb0ada89fbba54020c618907425dc858a2591b)
![[R] = {\ fra с {(1-ее) (1-с)} {2}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/1a61e8391de522b72a3580bd1214d5112057d741)
, где ee - это ee исходного материала. Обратите внимание, что для c = 0, что означает начало реакции, 
![{\ frac {d [S]} {dt}} = - k_ {S} [S] [B ^ {* }] \ подразумевает \ log [S] = - k_ {S} [B ^ {*}] t + \ log S_ {0}](https://wikimedia.org/api/rest_v1/media/math/render/svg/0cb65f6ee0307b80ab311cb9d09726947bd97ac1)
Обратите внимание, что если разделяющий агент стехиометрический и ахиральный, с хиральным катализатором, член [B *] не появляется. Тем не менее, используя такое же выражение для R, мы могли бы выразить s как
![{\ displaystyle s = {\ frac {k_ {R}} {k_ {S}}} = {\ frac {\ log [R] - \ log R_ {0}} {\ log [S] - \ log S_ {0}} } = {\ frac {\ log [(1-c) (1-ee)] + \ log {\ frac {1} {2}} - \ log R_ {0}} {\ log [(1-c) (1 + ee)] + \ log {\ frac {1} {2}} - \ log S_ {0}}} = {\ frac {\ log [(1-c) (1-ee)]} {\ log [(1-c) (1 + ee)]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/156b26edecc36af36374614253bb2842be3e550b)
Если мы хотим выразить это с точки зрения энантиомерного избытка продукта, ее ", мы должны использовать тот факт, что для продуктов R 'и S' из R и S, соответственно,
![ee''={\frac {[R']-[S']}{[R']+[S']}}={\frac {ee(1-c)}{c}}\implies ee=ee''{\frac {c}{1-c}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/0876f7366b97d07d9a002cb750fdb1a42ca25448)
Отсюда, мы видим, что


который дает нам


, когда мы подставляем в нашем выражении для s, полученное выше, получаем
![{\displaystyle s={\frac {\log[1-c(1+ee'')]}{\log[1-c(1-ee'')]}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/6d142162d7ab054c445d0ee122e6ed2b2880103a)
Кроме того, выражения для c и ee можно параметризовать для получения явных выражений для C и ee в терминах t. Во-первых, явное решение для [S] и [R] как функции от t дает
![{\ frac {d [S]} {dt}} = - k_ {S} [S] \ подразумевает S = {\ frac {1} {2}} e ^ {{ -k_ {S} t}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/8fcf79858e21ac5c20f56338e038c1a28e2b6e77)
, вставленный в выражении для ee и c дает
![ee = {\ frac {[S] - [R]} {[S] + [R]}} = {\ frac {e ^ {{- k_ {S} t}}} - e ^ {{- k_ {R} t}}} {e ^ {{- k_ {S} t}} + e ^ {{- k_ {R} t}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/537dcb1d65d95eea71ade3ed04fb39937e993fb2)
![c = 1 - {\ big (} [S] - [R] {\ big)} = 1 - {\ frac {e ^ {{- k_ {S} t}} + e ^ { {-k_ {R} t}}} {2}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/002346d2f288670c618bfcddfabc48db5fe6a46b)
Без ограничения общности мы можем допустить, что k S = 1, что дает k R = s, упрощенная приведенные выше выражения. Аналогичным выражением для ee ″ как функции от t может быть получено

Таким образом, графики ee и ee ″ в зависимости от c могут быть сгенерированы с t в качестве параметра и разными значениями s, генерирующими разными кривыми, как показано ниже.
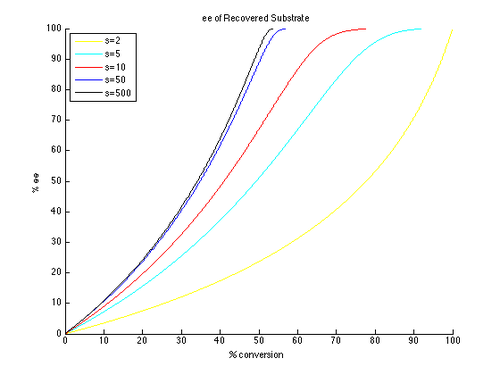
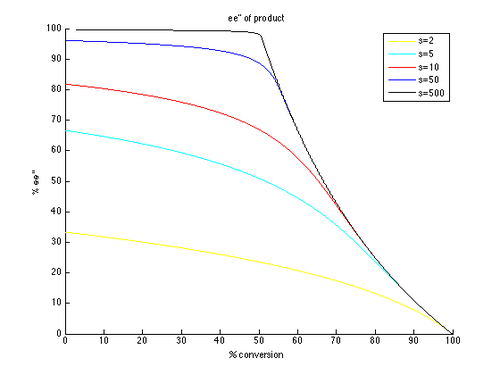
Как можно видеть, высокие энантиомерные избытки легче достижимы для непрореагировавшего исходного материала. Однако высокий ее (восстановленного субстрата) получается при более высокой конверсии и, следовательно, более низкий изолированного. Например, при коэффициенте селективности всего 10 возможно 99% ее при конверсии примерно 70%, что дает выход примерно 30%. Напротив, для получения хорошего э.и.и выхода продукта необходимы очень высокие коэффициенты селективности. Например, при коэффициенте селективности 10 ee ″ выше приблизительно 80% недостижимо, а для более реалистичных преобразований получаются значительно более низкие значения ee ″. Селективность выше 50 требуется для высокоэнантиообогащенного продукта с разумным выходом.
Следует отметить, что это упрощенная версия истинной кинетики кинетического разрешения. Предположение, что реакция является первым порядком по субстрату, является ограничивающим, и возможно, что зависимость от субстрата может зависеть от конверсии, что приводит к более сложной картине. В общем подходе содержится в том, чтобы измерять и сообщать только урожайность и ее, поскольку формула для k rel применяется только к идеализированному кинетическому разрешению. Несложно представить себе начальное образование комплекса субстрат-катализатор, которое может свести на нет кинетику первого порядка. Однако сделанные общие выводы по-прежнему полезны для понимания селективности и преобразования на ее.
С появлением асимметричного катализа необходимо рассмотреть практичность использования кинетического разрешения для получения энантиочистых продуктов. Даже для продукта, который может быть получен с помощью асимметричного каталитического или вспомогательного пути, рацемат может быть значительно дешевле, чем энантиочистый материал, что приводит к повышенной рентабельности даже с присущей «потерей» 50% материала. В качестве необходимых условий для практического кинетического разрешения были предложены следующие условия:
На сегодняшний день разработан ряд катализаторов кинетического разделения, которые удовлетворяют большинству, если не используют все из вышеперечисленныхев, что делает их делает их очень практичными для использования в органическом синтезе. В следующих разделах будет обсуждаться ряд основных примеров.
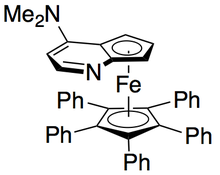 Плоский хиральный DMAP Фу (-) - катализатор кинетического разделения вторичных спиртов
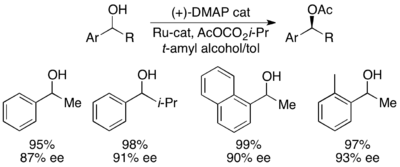
Плоский хиральный DMAP Фу (-) - катализатор кинетического разделения вторичных спиртов Грегори Фу и его коллеги разработали методологию с использованием хирального аналога DMAP для достижения превосходного кинетического разрешения вторичных спиртов. Первоначальные исследования с использованием простого эфира в качестве растворителя, низкого катализатора загрузки (2 мол.%), уксусного ангидрида в качестве ацилирующего агента и триэтиламина при комнатной температуре дали селективность в диапазоне от 14 до 52, что соответствует ее значению для восстановленного спиртового продукта 99,2%. Однако скрининг растворителей показал, что использование трет-амилового спирта увеличивает реакционную способность, так и селективность.

С эталонным субстратом 1-фенилэтанолом это соответствует 99% ее содержания непрореагировавшего спирта при 55% преобразовании при работе при 0 ° C. Эта система оказалась способной к разделению ряда арилалкилкарбинолов с селективностью до 95 и низкой загрузкой катализатора 1%, как показано ниже, с использованием (-) - энантиомера катализатора. Это к получению высокоэнантиообогащенных спиртов с очень низкой степенью привело конверсии, что также дало превосходные выходы. Кроме того, высокая селективность приводит к получению высокоэнантиообогащенных ацилированных продуктов с 90% э.и. пробы ацилированного спирта для о-толилметилкарбинола с s = 71.
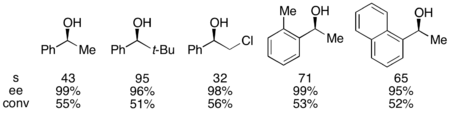
Кроме того, Fu сообщил о первом высокоселективном ацилировании рацемических диолов (а также о десимметризации мезодиолов). При низкой загрузке катализатора, составляющей 1%, энантиообогащенный диол был извлечен с его выходом 98% и выходом 43%, а диацетат - с выходом 39% и выходом 99%. Остаток материала извлекали в виде смеси моноацетата.

Планарно-хиральный катализатор DMAP также показал свою эффективность при кинетическом разделении пропаргиловых спиртов. Однако в этом случае селективность оказалась самой высокой без присутствия основания. При работе с 1 мол.% Катализатора при 0 ° C можно было достичь селективности до 20. Ограничение этого метода включает требование наличия ненасыщенной функциональной группы, такой как карбонил или алкены, в удаленном алкинильном положении. Спирты, разделенные с использованием (+) - энантиомера катализатора DMAP, показаны ниже.

Фу также применил способность своего хирального катализатора DMAP разделять аллильные спирты. Эффективная селективность зависела от наличия геминального или цис-заместителя у спиртосодержащей группы, за заметным исключением транс-фенилового спирта, который проявлял наивысшую селективность. Используя 1-2,5 мол.% (+) - энантиомера катализатора DMAP, спирты, показанные ниже, были разделены в присутствии триэтиламина.

 Катализатор Fu (-) - PPY * (слева) и новый ацилирующий агент (справа)
Катализатор Fu (-) - PPY * (слева) и новый ацилирующий агент (справа) Хотя катализатор-аналог DMAP Fu работал исключительно хорошо для кинетического разделения рацемических спиртов, его не удалось использовать для кинетического разделения аминов. Был разработан аналогичный катализатор PPY *, который при использовании с новым ацилирующим агентом удалось успешно провести ацилирование аминов с кинетическим разрешением. При 10 мол.% (-) - PPY * в хлороформе при –50 ° C наблюдалась селективность от хорошей до очень хорошей при ацилировании аминов, как показано ниже. Аналогичный протокол разработан для кинетического разделения индолинов.
Метод эпоксидирования Шарплесса, нас К. Барри Шарплесс в 1980 г. был использован для кинетического разделения рацемической смеси аллиловых спиртов. Хотя этот метод эффективен при разделении ряда аллиловых спиртов, он имеет ряд недостатков. Время реакции может достичь 6 дней, и катализатор не подлежит переработке. Тем не менее, кинетическое разрешение асимметричного эпоксидирования по Шарплессу остается одним из самых эффективных синтетических кинетических разрешений на сегодняшний день. В качестве катализатора можно использовать ряд различных тартратов; Ниже представлена типичная схема с использованием диизопропилтартрата. Этот метод нашел широкое применение для ряда вторичных аллильных спиртов.

Асимметричное дигидроксилирование по Шарплесу также было использовано в качестве метода кинетического разрешения. Однако этот метод широко не используется, поскольку одно и то же разрешение может быть достигнуто другими способами, которые являются более экономичными. Кроме того, было показано, что эпоксидирование Ши влияет на кинетическое разрешение ограниченного набора олефинов. Этот метод также не получил широкого распространения, но представляет механистический интерес.
 Катализатор Якобсена (R, R) (сален) -Cr для кинетического разделения концевых эпоксидов посредством образования азидов
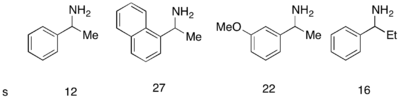
Катализатор Якобсена (R, R) (сален) -Cr для кинетического разделения концевых эпоксидов посредством образования азидов Хотя энантиоселективное эпоксидирование было успешно достигнуто с использованием эпоксидирования Шарплесса, Ши эпоксидирование и эпоксидирование Якобсена ни один из этих методов не позволяет обеспечить эффективный асимметричный синтез концевых эпоксидов, которые являются ключевыми хиральными строительными блоками. Эффективное кинетическое разделение терминальных эпоксидов могло бы служить очень синтетическим методологией. В 1996 году Якобсен и его коллеги разработали методологию кинетического разделения эпоксидов нуклеофильного раскрытия кольца с атакой азид-анионом. Показан катализатор (R, R). Катализатор может эффективно при загрузке всего 0,5 мол.% Энантиоселективно раскрывать эпоксид в концевом положении, давая энантиообогащенный исходный эпоксидный материал и 1,2-азидоспирты. Урожайность почти количественная, а ее превосходные (≥95% почти во всех случаях). Аминоспирты можно гидрогенизировать с получением 1,2-аминоспиртов, как показано ниже.
 Катализатор Якобсена (R, R) (сален) -Cr для гидролитического кинетического разделения концевых эпоксидов
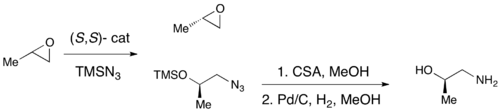
Катализатор Якобсена (R, R) (сален) -Cr для гидролитического кинетического разделения концевых эпоксидов В 1997 году группа Якобсена опубликовала методологию, которая улучшила их более ранние работы, позволив использовать воду в нуклеофила в эпоксидное отверстие. При использовании почти идентичного катализатора наблюдали ее более 98% как для извлеченного исходного эпоксида, так и для продукта 1,2-диола. В приведенном ниже примере гидролитическое кинетическое разрешение (HKR) было проведено в масштабе 58 грамм, что дало 26 г (44%) энантиорированного эпоксида в>99% ee и 38 г (50%) диола в 98%. ее.
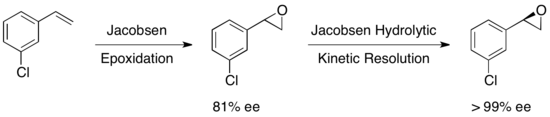
Было исследовано множество других субстратов с выходом восстановленного эпоксида в диапазоне от 36 до 48% до>99% ee. Гидролитическое кинетическое разрешение Якобсена можно использовать в тандеме с эпоксидированием Якобсена для получения энантиочистых эпоксидов из определенных олефинов, как показано ниже. Первое эпоксидирование дает слегка энантиообогащенный эпоксид, а последующее кинетическое разделение дает по существу единственный энантиомер. Преимущество этого подхода заключается в возможности уменьшить количество гидролитического расщепления, необходимого для достижения высокой энантиоселективности, что позволяет достичь общих выходов примерно до 90% в пересчете на олефин.
В конечном итоге кинетическое разрешение раскрытия эпоксида Якобсена дает высокие энантиомерные показатели. чистота эпоксида и продукта в условиях отсутствия растворителя или с низким содержанием растворителя, и были применимы в большом масштабе. В частности, метод Якобсена для HKR чрезвычайно привлекателен, поскольку он может быть реализован в многотонном масштабе и использует воду в качестве нуклеофила, что приводит к чрезвычайно рентабельным промышленным процессам. Несмотря на впечатляющие достижения, HKR обычно применялся для разрешения простых конечных эпоксидов с одним стереоцентром. Совсем недавно Д. А. Деваланкар и соавт. сообщили об элегантном протоколе, включающем два стереоцентрированных сокатализируемого HKR рацемических концевых эпоксидов, несущих соседние C – C-связывающие заместители.
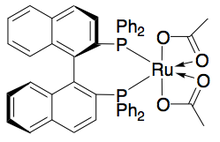
 Катализатор Нойори (S, S) для гидрогенизации с переносом / кинетического разрешения вторичныеспирты
Катализатор Нойори (S, S) для гидрогенизации с переносом / кинетического разрешения вторичныеспирты Ryōji Noyori и его коллеги разработали методологи кинетического разделения бензиловых и аллильных вторичных спиртов посредством гидрогенизации с переносом. Комплекс рутения катализирует окисление более реакционноспособного энантиомера из ацетона с образованием непрореагировавшего энантиочистого спирта, окисленного кетона и изопропанола. В примере проиллюстрированного ниже воздействия на 1-фенилэтанол (S, S) энантиомера катализатора в присутствии ацетона дает выход 51% для 94% ее (R) -1-фенилэтанола вместе с 49% ацетофенона. и изопропанол в побочном продукте.
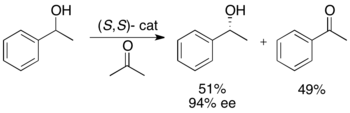
Эта методология, по существу, является обратной методике гидрирования кетонов асимметричным способом, предложенной Нойори, в результате восстановления получаются энантиообогащенные спирты. Это ограничивает использование метода кинетического разрешения, поскольку существует аналогичный метод получения тех же продуктов без потери материала. Таким образом, кинетическое разделение можно было бы проводить только в том случае, когда рацемический спирт был по крайней вдвое дешевле кетона или был значительно более доступным.

 Катализатор Уэмура и Хидаи для гидрогенизации с переносом / кинетического разделения катализаторов
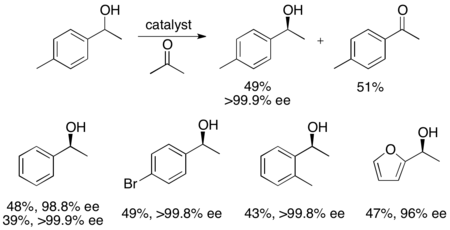
Катализатор Уэмура и Хидаи для гидрогенизации с переносом / кинетического разделения катализаторов Кроме того, Уэмура и Хидаиали рутениевый катализатор для окисления бензиловых спиртов с кинетическим разрешением, дающий высокоэнантиообогащенные спирты с хорошими выходами. Комплекс может, как катализатор Нойори, влиять на гидрогенизацию переноса между кетоном и изопропанолом с образованием энантиообогащенного спирта, а также влиять на кинетическое разрешение рацемического спирта, давая энантиоченный спирт (>99%) и окисленный кетон с ее ацетоном в побочном продукте.. Он очень эффективен при энантиоселективном восстановлении кетонов, давая большинство бензиловых спиртов с э.и.>99% и может растворять ряд рацемических бензиловых спиртов с получением выходов (до 49%) отдельных энантиомеров, как показано ниже. Этот метод имеет те же недостатки, что и кинетическое разрешение Нойори, что к спиртам можно также получить доступ посредством энантиоселективного восстановления кетонов. Кроме того, сообщалось только об одном энантиомере катализатора.
 Катализатор Noyori (R) -BINAP Ru для гидрогенизирующего кинетического разделения аллиловых спиртов
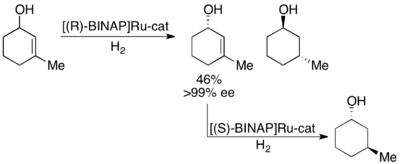
Катализатор Noyori (R) -BINAP Ru для гидрогенизирующего кинетического разделения аллиловых спиртов Noyori также использует кинетическое разделение аллиловых спиртов путем асимметричного гидрирования олефина. Используя комплекс Ru [BINAP], может селективное гидрирование дать высокие ее значения ненасыщенного спирта в дополнение к гидрированному спирту, как показано ниже. Таким образом, второе гидрирование оставшегося энантиообогащенного аллилового спирта даст энантиомерно чистые образцы обоих энантиомеров насыщенного спирта. Нойори разделил ряд аллиловых спиртов с выходом от хорошего до отличного и от хорошего до отличного (до>99%).
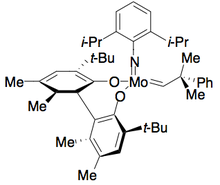 Катализатор Ховейды и Шрока для кинетического разрешения метатезиса с замыканием цикла
Катализатор Ховейды и Шрока для кинетического разрешения метатезиса с замыканием цикла Ховейда и Шрок разработали катализатор для метатезиса с замыканием цикла кинетического разделения диенилаллических спиртов. Катализатор молибден алкилиден селективно катализирует один энантиомер для осуществления метатезиса замыканием цикла, в результате чего получают энантиочистый спирт и энантиочистое замкнутое кольцо, как показано ниже. Катализатор наиболее эффективен при разделении 1,6-диенов. Однако небольшие структурные изменения в субстрате, такие как увеличение расстояния между алкенами до 1,7, могут потребовать использования другого катализатора, что снижает эффективность этого метода.

Как и процедуры синтетического кинетического разрешения, ферментативное кинетическое разрешение ацилирования нашло самое широкое применение в синтетическом контексте. Особенно важно было использование ферментативного кинетического разрешения для эффективного и дешевого использования аминокислот. В коммерческих масштабах методология Degussa с использованием ацилаз позволяет разделить множество природных и неприродных аминокислот. Растворимые смеси могут быть приготовлены с помощью синтеза Штрекера, и использование ацилазы почекьи (для субстратов с прямым цепью) или фермента из плесени Aspergillus oryzae (для субстратов с разветвленной боковой цепью) может эффективно применять энантиообогащенные аминокислоты с высокими (85-90%) выходами. Непрореагировавший исходный материал может быть рацемизирован in situ, что обеспечивает динамическое кинетическое разрешение.

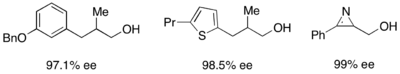
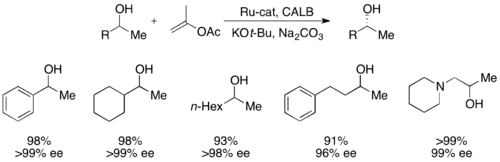
Кроме того, липазы широко используются для кинетического разрешения как в академических, так и в промышленных условиях. Липазы использовались для разделения первичных спиртов, вторичных спиртов, ограниченного числа третичных спиртов, карбоновых кислот, диолов и даже хиральных алленов. Липаза из Pseudomonas cepacia (PSL) наиболее широко используется для разделения первичных спиртов и используется с винилацет в качестве ацилирующего агента кинетического разделения первичных спиртов, показанных ниже.
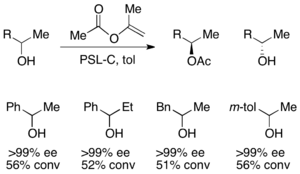
Для разделения вторичных спиртов липаза pseudomonas cepecia (PSL-C) была примен для превосходного ее (R) -энантиомера спирта. Использование изопропенилацетата в качестве ацилирующего агента приводит к ацетону в побочном продукте, который эффективно использует реакции с использованием молекулярных сит.
Пекарские дрожжи (BY) были использованы для кинетического разрешения α- стереогенных карбонильных соединений. Фермент избирательно восстанавливает один энантиомер, давая высокоэнантиообогащенный спирт и кетон, как показано ниже.

Пекарские дрожжи также применялись для кинетического разделения бензиловых спиртов путем окисления. Несмотря на то, что сообщалось об отличном э.и. регенерированного спирта, для них обычно требуется конверсия>60%, что приводит к снижению выхода. Пекарские дрожжи также использовались для кинетического метода восстановления β-кет разрешенияоэфиров. Однако, учитывая статью успешное разрешение Нойори тех же субстратов, подробно описанное далее в этой статье.
Динамическое кинетическое разрешение (DKR) происходит, когда рацематический материал способен легко эпимеризоваться, что приводит к по существу рацемической смеси материалов во всех точках во время реакции. Тогда энантиомер с более низким барьером активации может образовываться с теоретическим выходом до 100%. Это показывает со стандартным кинетическим выходным разрешением, которое обязательно имеет максимальный 50%. По этой причине динамическое кинетическое разрешение имеет практическое применение в органическом синтезе. Наблюдаемая основа на принцип Куртина-Хэммета. Барьер реакции любого энантиомера обязательно выше, чем барьер эпимеризации, в результате чего кинетическая яма содержит рацемат. Это эквивалентно написано для k R>kS,


Был опубликован ряд превосходных обзоров, последний из которых в 2008 г., в которых подробно описаны теория и практическое применение DKR.
Асимметричное гидрирование Нойори кетонов является прекрасным примером динамического кинетического разрешения в действии. Энантиомерные β-кетоэфиры могут подвергаться эпимеризации и выбору хирального катализатора, обычно в форме Ru [(R) -BINAP] X 2, где X представляет собой галоген, приводит к тому, что один из энантиомеров реагирует быстрее. Относительная свободная энергия для типичной реакции ниже. Как можно, промежуточное соединение эпимеризации имеет более низкую свободную энергию, чем переходные состояния для гидрирования, что приводит к быстрой рацемизации и высокому выходам одного энантиомера продукта.
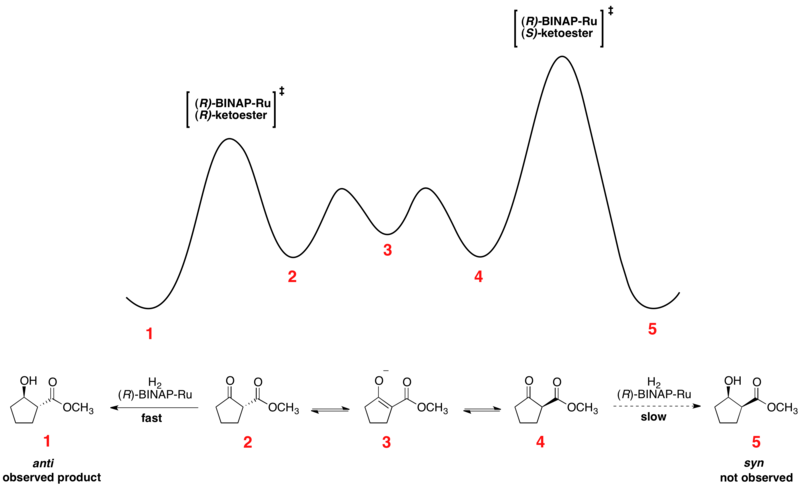
Энантиомеры взаимопревращаются посредством их общего енола, который представляет собой энергетический минимум, расположенный между энантиомерами. Показанная реакция дает 93% -ный образец анти-продукта, показанного выше. Выбор растворителя, по-видимому, имеет большое влияние на диастереоселективность, поскольку и дихлорметан, и метанол проявляют эффективность для определенных субстратов. Нойори и другие также разработали новые катализаторы, которые улучшили ее, так и диастереомерное соотношение (dr).
 (S) -SYNPHOS
(S) -SYNPHOS Генет и его сотрудники разработали SYNPHOS, аналог BINAP, который образует комплексы рутения, которые обеспечивают высокоселективное асимметричное гидрирование. Было показано, что Enantiopure Ru [SYNPHOS] Br 2 селективно гидрирует рацемические α-амино-β-кетоэфиры до энантиочистых аминоспиртов, как показано ниже, с использованием (R) -SYNPHOS. 1,2-синаминоспирты предлагают из бензоил защищенных аминосоединений, тогда как анти-продукты существуют из гидрохлоридных солей амина.
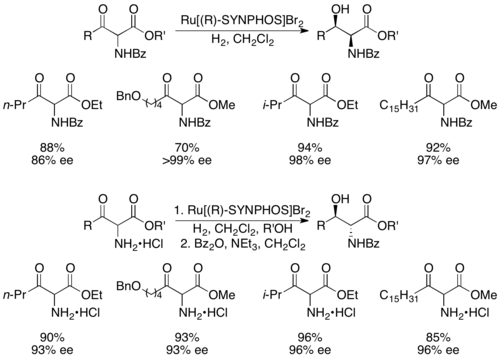
 Рутениевый катализатор, инструмент Fu для рацемизации вторичных спиртов
Рутениевый катализатор, инструмент Fu для рацемизации вторичных спиртов Недавно Грегори Фу и его коллеги сообщили о модификации своей более ранней работы по кинетическому разрешению, чтобы обеспечить эффективное динамическое кинетическое разрешение. Используя катализатор рацемизации рутения, показанный справа и его планарный хиральный катализатор DMAP, используйте динамическое кинетическое разрешение вторичных спиртов с выходом до 99% и 93% ее, как показано ниже. Продолжаются работы по дальнейшему развитию приложений широко используемого катализатора DMAP для динамического кинетического разрешения.
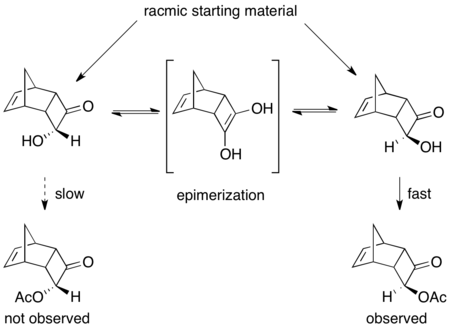
Сообщалось о ряде ферментативных динамических кинетических разрешений. Первичный пример использования PSL эффективно использует рацемические ацилоины в присутствии триэтиламина и винилацетата в качестве ацилирующего агента. Как показано ниже, продукт был выделен с выходом 75% и ee 97%. Без присутствия основания происходило регулярное кинетическое разрешение, приводящее к выходу 45% ацилированного продукта>99% ее и 53% исходного материала с 92% ее.
Другим превосходным, хотя и не очень продуктивным примером является кинетическое разрешение (±) -8-амино-5,6,7,8-тетрагидрохинолина. При воздействии липазы B Candida antarctica (CALB) в толуоле и этилацетате в течение 3–24 часов происходит нормальное кинетическое разрешение, что приводит к выходу 45% или 97% ее исходного материала. и 45% выход продукта ацилированного амина>97% ее. Однако, когда реакционная смесь дает возможность перемешиваться в течение 40-48 часов, выделяется рацемический исходный материал и>60% ацилированного продукта>95%.
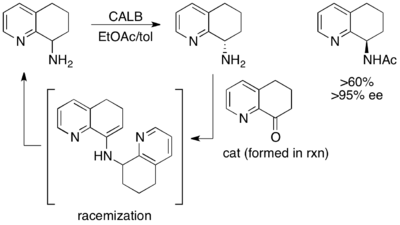
Здесь непрореагировавший исходный материал рацемизируется in situ через димерный енамин, в результате чего при извлечении более чем 50% выхода энантиочистого ацилированного аминного продукта.
Имеется ряд описанных процедур, в которых используются преимущества химического реагента / катализатора для проведения рацемизации исходного материала и фермента для селективной реакции с одним энантиомером, называется хемоэнзиматическим динамическим кинетическим разрешением. PSL-C использовался вместе с рутениемвым катализатором (для рацемизации) для получения энантиочистых (>95% ее) δ-гидроксилактонов.
 Рутениевый катализатор Бэквалла для рацемизации в хемоэнзиматическом динамическом кинетическом разделении вторичного спирта
Рутениевый катализатор Бэквалла для рацемизации в хемоэнзиматическом динамическом кинетическом разделении вторичного спирта Позже вторичные спирты спирты были разделены Bäckvall с выходами до 99% и ее до>99% с использованием CALB и комплекса рацемизации рутения.
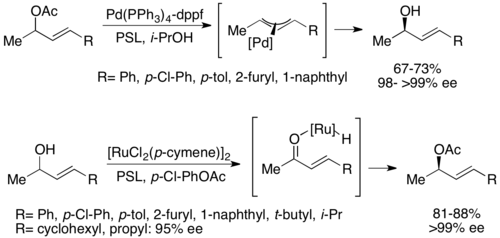
Второй тип химико-ферментативного динамического кинетического разрешения включает π-аллильный комплекс из аллильного ацетата с палладий. Здесь рацемизация происходит с потерей ацетата с образованием катионного комплекса с переходным металлом, как показано ниже. Было показано, что палладий способствует этой реакции, в то время как рутений влияет на аналогичную реакцию, также показанную ниже.
При параллельном кинетическом разрешении (PKR) рацемическая смесь реагирует с образованием два неэнантиомерных продукта, часто совершенно разными путями реакции. С PKR нет компромисса между конверсией и ее, так как образующиеся продукты не являются энантиомерами. Одна стратегия PKR заключается в удалении менее реакционноспособного энантиомера (по отношению к желаемому хиральному катализатору) из реакционной смеси, подвергая его второму набору условий реакции, которые предпочтительно реагируют с ним, в идеале с примерно равной скоростью реакции. Таким образом, оба энантиомера потребляются разными путями с одинаковой скоростью. Эксперименты PKR могут быть стереодивергентными, региодергентными или структурно расходящимися. Один из наиболее эффективных PKR, о котором сообщалось на сегодняшний день, был выполнен Йошито Киси в 1998 году; CBS-восстановление рацемического стероидного кетона привело к стереоселективному восстановлению с образованием двух диастереомеров с>99% ее, как показано ниже.

PKR также была достигнута с использованием ферментных катализаторов. При использовании гриба Mortierella isabellina NRRL 1757 восстановление рацемических β-кетонитрилов дает два диастереомера, которые можно разделить и повторно окислить с получением высокоэнантиочистых β-кетонитрилов. Однако в высшей степени синтетически полезные параллельные кинетические разрешения еще предстоит открыть. Был обнаружен ряд процедур, которые дают приемлемые ЭИ и выходы, но очень мало примеров, которые дают высокоселективное параллельное кинетическое разрешение, а не просто несколько селективные реакции. Например, параллельное кинетическое разделение 4-алкиналов Фу дает очень энантиообогащенный циклобутанон с низким выходом и слегка энантиообогащенный циклопентенон, как показано ниже.

Теоретически параллельное кинетическое разрешение может дать самые высокие ее продуктов, поскольку только один энантиомер дает каждый желаемый продукт. Например, для двух взаимодополняющих реакций с s = 49 конверсия 100% даст продукты с выходом 50% и ее 96%. Эти же значения потребуют s = 200 для простого кинетического разрешения. Таким образом, обещание PKR продолжает привлекать большое внимание. Сокращение Kishi CBS остается одним из немногих примеров выполнения этого обещания.