Войти
 Общая схема процесса Wacker
Общая схема процесса Wacker Процесс Wacker или процесс Hoechst-Wacker (названный в честь одноименных химических компаний) относится к окислению этилена до ацетальдегида в присутствии хлорида палладия (II) как катализатор. Эта химическая реакция была одной из первых гомогенного катализа с применением химии палладия в промышленных масштабах.
The Wacker Впервые о реакции сообщили Смидт и др.
Разработка химического процесса, ныне известного как процесс Wacker, началась в 1956 г. в Wacker Chemie. В то время многие промышленные соединения производились из ацетилена, полученного из карбида кальция, - дорогостоящей и экологически вредной технологии. Строительство нового нефтеперерабатывающего завода в Кельне компанией Esso рядом с производственной площадкой Wacker в сочетании с осознанием того, что этилен будет более дешевым сырьем, побудило компанию Wacker для изучения его потенциального использования. В рамках последующих исследований реакция этилена и кислорода на палладий на углероде в поисках этиленоксида неожиданно дала свидетельства образования ацетальдегида (просто по запаху). Дальнейшие исследования превращения этилена в ацетальдегид привели к патенту 1957 года, описывающему газофазную реакцию с использованием гетерогенного катализатора. Тем временем Hoechst AG присоединилась к гонке и после подачи заявки на патент вынудила Wacker вступить в партнерство под названием Aldehyd GmbH . Гетерогенный процесс в конечном итоге потерпел неудачу из-за инактивации катализатора и был заменен гомогенной системой на водной основе, для которой пилотная установка была запущена в 1958 году. Проблемы с агрессивным раствором катализатора были решены за счет использования титана (недавно доступного для промышленное использование) в качестве конструкционного материала для реакторов и насосов. Производственные предприятия были введены в эксплуатацию в 1960 году.
Механизм реакции для промышленного процесса Ваккера (окисление олефинов через хлорид палладия (II)) привлек значительное внимание. в течение нескольких десятилетий. Аспекты механизма все еще обсуждаются. Современный состав описан ниже:
 Каталитический цикл для процесса Ваккера.
Каталитический цикл для процесса Ваккера. Первоначальная стехиометрическая реакция была впервые описана Филлипсом. Чистую реакцию также можно описать следующим образом:
За этим превращением следуют реакции, которые регенерируют катализатор Pd (II):
Расходуются только алкен и кислород. Без хлорида меди (II) в качестве окислителя, металлический Pd (0) (полученный в результате отщепления бета-гидрида Pd (II) на последней стадии) будет выпадать в осадок, останавливая реакцию после одного цикла. Эта стехиометрическая реакция была открыта в 1894 году. Воздух, чистый кислород или ряд других реагентов могут затем окислить образовавшуюся смесь CuCl -хлорид обратно до CuCl 2, позволяя циклу продолжаться..
Ранние механистические исследования 1960-х годов прояснили несколько ключевых моментов:
Многие механистические исследования процесса Ваккера были сосредоточены на пути образования связи CO, стадии гидроксипалладирования . Генри пришел к выводу, что скоординированный гидроксид атакует этиленовый лиганд, внутренний (син-) путь. Позже стереохимические исследования Стилла с соавторами подтверждают наличие пути против присоединения, посредством которого свободный гидроксид атакует этиленовый лиганд. Условия экспериментов Стилле существенно отличаются от условий промышленного процесса. Другие исследования с использованием нормальных промышленных условий Ваккера (за исключением высоких концентраций хлоридов и высоких концентраций хлорида меди) также дали продукты, предполагающие, что нуклеофильная атака является реакцией против присоединения.
Кинетические исследования были проведены на изотопно замещенных аллиловых спиртах в стандартных промышленных условиях. условия (с низкими концентрациями хлоридов), чтобы исследовать механизмы реакции. Эти результаты показали, что нуклеофильная атака - это медленный процесс, в то время как предложенные механизмы, объясняющие более ранние стереохимические исследования, предполагали, что нуклеофильная атака является быстрым процессом.
Последующие стереохимические исследования показали, что оба эти пути возникают и зависят от концентраций хлоридов. Однако эти исследования также оспариваются, поскольку аллиловые спирты могут быть чувствительны к реакциям изомеризации, и различные стереоизомеры могут образовываться из этих реакций, а не из стандартного процесса Ваккера.
Таким образом, экспериментальные данные, по-видимому, подтверждают, что син-присоединение происходит при низких концентрациях хлорида в реакции (<1 моль /L, в условиях промышленного процесса), в то время как анти-присоединение происходит при высоком содержании хлоридов. (>3 моль /L ) реакционных концентраций, вероятно, из-за насыщения катализатора ионами хлора и ингибирования механизма внутренней сферы. Однако точный путь и причина этого переключения путей до сих пор неизвестны.
Еще больше усложняют механизм процесса Ваккера вопросы о роли хлорида меди. Большинство теорий предполагало, что медь не играет роли в механизмах окисления олефинов. Тем не менее, эксперименты Штангла и Джира показали, что образование хлоргидрина зависит от концентрации хлорида меди. Работа Хосокавы и его сотрудников позволила получить кристаллизованный продукт, содержащий хлорид меди, что указывает на то, что он может играть небезопасную роль в окислении олефинов. И, наконец, исследование, проведенное с первых шагов Comas-Vives и соавт. с участием медного сокатализатора не обнаружено, что антидобавление является предпочтительным путем. Этот путь позже был подтвержден экспериментами Андерсона и Сигмана без содержания меди. Другой закон кинетической скорости без протонной зависимости был обнаружен в условиях отсутствия меди, что указывает на возможность того, что даже небольшие количества медных сокатализаторов могут играть небезопасную роль в этой химии. Хотя эти работы усложняют картину механизма процесса Ваккера, следует, вероятно, сделать вывод, что этот и связанный с ним химический состав могут быть чувствительны к условиям реакции, и могут быть задействованы несколько различных путей реакции.
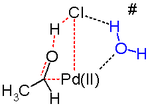
Другим ключевым этапом процесса Wacker является миграция водорода из кислорода в хлорид и образование двойной связи C-O. Обычно считается, что на этом этапе происходит так называемое отщепление β-гидрида с циклическим четырехчленным переходным состоянием :
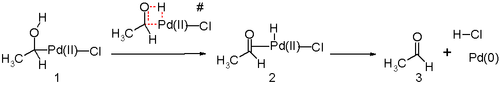
In silico. Исследования показывают, что переходное состояние для этой стадии реакции является неблагоприятным, и действует альтернативный механизм реакции восстановительного отщепления . Предложенным стадиям реакции, вероятно, способствует молекула воды в растворе, действующая как катализатор.
Для производства ацетальдегида коммерциализируются два направления: одностадийный процесс и двухстадийный.
Этилен и кислород одновременно пропускаются в реакционной башне при примерно 130 ° C и 400 кПа. Катализатор представляет собой водный раствор PdCl 2 и CuCl 2. Ацетальдегид очищают с помощью экстрактивной дистилляции с последующей фракционной перегонкой. Экстракционная дистилляция с водой удаляет легкие фракции с более низкими точками кипения, чем у ацетальдегида (хлорметан, хлорэтан и диоксид углерода ) вверху, а вода и выше - побочные продукты кипения, такие как уксусная кислота, кротоновый альдегид или хлорированные ацетальдегиды, удаляются вместе с ацетальдегидом на дне. Из-за коррозионной природы катализатора, реактор облицован кислотостойким керамическим материалом, а трубки изготовлены из титана.
В двухстадийном процессе реакция и окисление осуществляются отдельно в трубчатых реакторах. В отличие от одностадийного процесса вместо кислорода можно использовать воздух. Этилен пропускается через реактор вместе с катализатором при 105–110 ° C и 900–1000 кПа. Раствор катализатора, содержащий ацетальдегид, отделяют с помощью мгновенной перегонки. Катализатор окисляется в реакторе окисления при 1000 кПа с использованием воздуха в качестве окислительной среды. Окисленный раствор катализатора отделяют и отправляют обратно в реактор. Кислород из воздуха полностью расходуется, а отработанный воздух циркулирует как инертный газ. Смесь ацетальдегид - водяной пар предварительно концентрируется до 60–90% ацетальдегида за счет использования тепла реакции, и сбрасываемая вода возвращается в испарительную колонну для поддержания концентрации катализатора. Далее следует двухступенчатая перегонка сырого ацетальдегида. На первой стадии отделяются низкокипящие вещества, такие как хлорметан, хлорэтан и диоксид углерода. На второй стадии удаляют воду и высококипящие побочные продукты, такие как хлорированный ацетальдегид и уксусная кислота, и получают ацетальдегид в чистом виде с головным погоном. Из-за коррозионной природы катализатора, оборудование, контактирующее с ним, футеровано титаном.
. Как в одно-, так и в двухстадийных процессах выход ацетальдегида составляет около 95%, а производственные затраты практически одинаковы. Преимущество использования разбавленных газов в двухступенчатом методе уравновешивается более высокими инвестиционными затратами. Оба метода дают хлорированные углеводороды, хлорированные ацетальдегиды и уксусную кислоту в качестве побочных продуктов. Как правило, выбор метода зависит от сырьевых материалов и энергии, а также от доступности кислорода по разумной цене. Обычно из 100 частей этилена получают:
и других второстепенных побочных продуктов

Поток диаграмма, показывающая технологическую схему одностадийного процесса Wacker для производства ацетальдегида.

Блок-схема, показывающая блок-схему двухстадийного процесса Wacker для производства ацетальдегида.
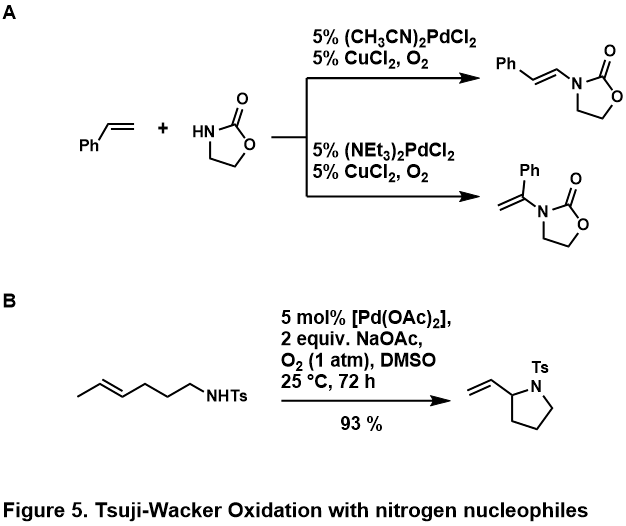
Появление Wacker Process стимулировало многие исследования полезности и применимости реакций к более сложным концевым олефинам. Окисление Цуджи-Ваккера представляет собой катализируемое палладием (II) превращение таких олефинов в карбонильные соединения. Клемент и Селвиц были первыми, кто обнаружил, что использование водного ДМФА в качестве растворителя позволяет окислять 1-додецен до 2-додеканона, что решает проблему нерастворимости олефинов более высокого порядка в воде. Фэи отметил, что использование 3-метилсульфолана вместо ДМФА в качестве растворителя увеличивает выход окисления 3,3-диметилбут-1-ена. Два года спустя Цуджи применил условия Зельвица для селективного окисления концевых олефинов с множеством функциональных групп и продемонстрировал их полезность в синтезе сложных субстратов. Дальнейшее развитие реакции привело к созданию различных каталитических систем для решения проблемы селективности реакции, а также к введению межмолекулярного и внутримолекулярного окисления с нуклеофилами, не являющимися водными.
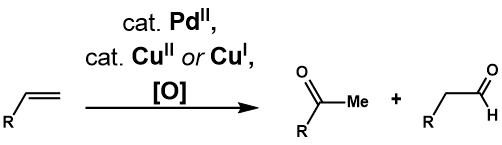
Окисление Цуджи-Ваккера окисляет концевой олефин до соответствующего метилкетона в условиях процесса Ваккера. Практически идентичный процессу Wacker Process, предлагаемый каталитический цикл (рисунок 1) начинается с комплексообразования PdCl 2 и двух хлорид-анионов до PdCl 4, которые затем подвергается последующей замене лиганда двух хлоридных лигандов на воду и алкен с образованием комплекса Pd (Cl 2) (H 2 O) (алкен). Затем молекула воды региоселективно атакует олефин через механизм внешней сферы по типу Марковникова с образованием более термодинамически стабильного Pd (Cl 2) (OH) (-CH 2-4 -CHOH-R) комплекс. Диссоциация хлоридного лиганда до трехкоординатного комплекса палладия способствует элиминированию β-гидрида, затем последующее введение 1,2-гидрида образует Pd (Cl 2) (OH) (-CHOHR-CH3 3) комплекс. Он подвергается элиминированию β-гидрида с высвобождением кетона, а последующее восстановительное элиминирование дает HCl, воду и палладий (0). Наконец, палладий (0) повторно окисляется до PdCl 2 двумя эквивалентами Cu (II) Cl 2, который, в свою очередь, может быть повторно окислен O 2.
. Обычно окисление концевых олефинов обеспечивают кетонный продукт Марковникова, однако в случаях, когда субстрат отдает предпочтение альдегиду (обсуждается ниже), для усиления региоселективности Марковникова можно использовать различные лиганды. Использование спартеина в качестве лиганда (рис. 2, A) способствует нуклеопалладированию на концевом атоме углерода для минимизации стерического взаимодействия между комплексом палладия и субстратом. Хинокс-лигированный палладиевый катализатор используется для содействия образованию кетона, когда субстрат содержит направляющую группу (рис. 2, B). Когда такой субстрат связывается с Pd (Quinox) (OOtBu), этот комплекс координационно насыщается, что предотвращает связывание направляющей группы и приводит к образованию марковниковского продукта. Эффективность этого лиганда также объясняется его электронным свойством, когда анионный ТБГП предпочитает связывать транс с оксазолином, а олефин координирует транс с хинолином.

Антимарковниковская селективность по альдегиду может быть достигнута за счет использования внутренней стереоэлектроники подложки. Размещение направляющей группы в гомо-аллильном (т.е. фиг. 3, A) и аллильном положении (т.е. фиг. 3, B) к концевому олефину способствует образованию антимарковниковского альдегидного продукта, который предполагает, что в каталитическом цикле направляющая группа хелатирует комплекс палладия, так что вода атакует антимарковниковский углерод с образованием более термодинамически стабильного палладоцикла. Антимарковниковская селективность также наблюдается в стиренильных субстратах (т.е. рис. 3, C), предположительно через комплекс η-палладий-стирол после того, как вода атакует антимарковниковые. Другие примеры субстрат-контролируемого анти-марковниковского окисления олефинов по Цуджи-Ваккеру приведены в обзорах Намбутири, Феринга и Музарта.
Граббс и его сотрудники проложили путь для анти-марковниковского окисления стереоэлектронно несмещенные концевые олефины за счет использования палладий-нитритной системы (рис. 2, D). В его системе концевой олефин был окислен до альдегида с высокой селективностью через катализатор-контролируемый путь. Механизм изучается, однако данные свидетельствуют о том, что он проходит через нитрит радикал, добавляемый к концевому углероду, с образованием более термодинамически стабильного вторичного радикала. Граббс расширил эту методологию на более сложные, несмещенные олефины.

Межмолекулярное окисление олефинов спиртами как нуклеофил обычно дает кетали, где в качестве катализируемого палладием окисления олефинов карбоновыми кислотами в качестве нуклеофилов образуются виниловые или аллильные карбоксилаты. В случае диолов их реакции с алкенами обычно образуют кетали, тогда как реакции олефинов, содержащих электроноакцепторные группы, имеют тенденцию к образованию ацеталей.
Катализируемое палладием межмолекулярное окисление диенов с карбоновыми кислотами и спиртами в качестве доноров дают продукты 1,4-присоединения. В случае циклогексадиена (рис. 4, А) Бэквалл обнаружил, что стереохимический выход продукта зависел от концентрации LiCl. Эта реакция протекает сначала путем образования комплекса Pd (OAc) (бензохинон) (аллил) путем антинуклеопалладирования диена с ацетатом в качестве нуклеофила. Отсутствие LiCl вызывает восстановительное отщепление внутренней сферы, что приводит к стереохимии транс-ацетата с образованием транс-1,4-аддукта. Присутствие LiCl замещает ацетат хлоридом из-за его более высокого сродства к связыванию, что заставляет ацетат внешней сферы атаковать палладий и обеспечивает цис-ацетатную стереохимию с образованием цис-1,4-аддукта. Внутримолекулярная окислительная циклизация: 2- (2-циклогексенил) фенол циклизуется до соответствующего дигидробензофурана (рис. 4, B); 1-циклогексадиен-уксусная кислота в присутствии уксусной кислоты циклизуется с образованием соответствующего аддукта лактон-ацетат 1,4 (фиг. 4, C) с селективностью цис и транс, контролируемой присутствием LiCl.
Окислительные аминирования олефинов обычно проводят с амидами или имидами ; Считается, что амины протонированы кислотной средой или слишком прочно связывают металлический центр, чтобы иметь место каталитическая химия. Было обнаружено, что эти азотные нуклеофилы компетентны как в межмолекулярных, так и в внутримолекулярных реакциях, приведены некоторые примеры (рис. 5, A, B)