Spartan - это молекулярное моделирование и вычислительная химия приложение от Wavefunction. Он содержит код для молекулярной механики, полуэмпирических методов, ab initio моделей, моделей функциональной плотности, пост-Хартри –Fock модели и термохимические рецепты, включая G3 (MP2) и T1. Расчеты квантовой химии в Spartan основаны на Q-Chem.
. Основные функции - предоставление информации о структурах, относительной стабильности и других свойствах изолированных молекул. Молекулярно-механические расчеты сложных молекул широко распространены в химическом сообществе. Квантово-химические расчеты, в том числе метод Хартри – Фока расчеты молекулярных орбиталей, но особенно расчеты, включающие электронную корреляцию, по сравнению с ними занимают больше времени.
Квантово-химические расчеты также используются для получения информации о механизмах и распределении продуктов химических реакций либо непосредственно путем расчетов переходных состояний, либо на основе постулата Хаммонда путем моделирования стерических и электронных требований к реагентам. Количественные расчеты, ведущие непосредственно к информации о геометрии переходных состояний и о механизмах реакций в целом, становятся все более распространенными, в то время как качественные модели по-прежнему необходимы для систем, которые слишком велики для подвергнуться более строгому обращению. Квантово-химические расчеты могут предоставить информацию, чтобы дополнить существующие экспериментальные данные или полностью заменить их, например, атомные заряды для количественного анализа зависимости структура-активность (QSAR) и межмолекулярные потенциалы для <128.>молекулярная механика и молекулярная динамика расчеты.
Spartan применяет методы вычислительной химии (теоретические модели) ко многим стандартным задачам, которые предоставляют расчетные данные, применимые для определения молекулярной формы конформации, структуры (равновесное и переходное состояние геометрия), ЯМР, ИК, Рамана и УФ-видимая спектры, молекулярные (и атомные) свойства, реакционная способность и селективность.
Содержание
- 1 Вычислительные возможности
- 2 Выполняемые задачи
- 3 Графический интерфейс пользователя
- 4 Графические модели
- 5 Спектральные вычисления
- 6 Базы данных
- 7 История основных выпусков
- 7.1 Версии Windows, Macintosh, Linux
- 8 См. Также
- 9 Ссылки
- 10 Внешние ссылки
Вычислительные возможности
Это программное обеспечение обеспечивает молекулярную механику, Merck Molecular Force Field (MMFF), (для набора проверочных тестов), MMFF с расширениями и SYBYL, расчет силовых полей, Полуэмпирические вычисления, MNDO / MNDO (D), Austin Model 1 (AM1), PM3, Recife Model 1 (RM1) PM6.
- Hartree– Fock, методы самосогласованного поля (SCF), доступные с неявным растворителем (SM8).
- методы теории функционала плотности (DFT), доступные с неявным растворителем (SM8).
- Стандартные функционалы: BP, BLYP, B3LYP, EDF1, EDF2, M06, ωB97X-D

Расчетная теплота образования T1 (ось y) относительно экспериментальной теплоты образования (ось x) для набора>1800 различных органических молекул из термохимической базы данных NIST со средним абсолютным и среднеквадратичным значением ошибки 8,5 и 11,5 кДж / моль соответственно.
- Функционалы обмена: HF, Слейтера-Дирака, Becke88, Gill96, GG99, B (EDF1), PW91
- Функционалы корреляции: VWN, LYP, PW91, P86, PZ81, PBE.
- Комбинированные или гибридные функционалы : B3PW91, B3LYP, B3LYP5, EDF1, EDF2, BMK
- Функционалы группы Трулара: M05, M05-2X, M06, M06-L M06-2X, M06-HF
- Групповые функционалы Хеда-Гордона: ωB97, ωB97X, ωB97X-D
- Методы связанных кластеров.
- CCSD, CCSD (T), CCSD (2), OD, OD (T), OD (2), QCCD, VOD, VOD (2), VQCCD
- Методы Меллера – Плессета.
- Методы возбужденного состояния.
- Композитные методы квантовой химии, термохимические рецепты.
Выполняемые задачи
Доступные вычислительные модели обеспечивают молекулярные, термодинамические, QSAR, атомные, графические и спектральные свойства. Диалог вычислений обеспечивает доступ к следующим вычислительным задачам:
- Энергия - для заданной геометрии предоставляет энергию и связанные с ней свойства молекулы или системы. Если используются квантово-химические модели, вычисляется волновая функция.
- Равновесие геометрия молекулы - определяет ближайший локальный минимум и обеспечивает энергию и связанные с ним свойства.
- Переходное состояние геометрия - определяет ближайшую седловую точку первого порядка (максимум в одном измерении и минимум во всех остальных) и обеспечивает энергию и соответствующие свойства.
- Конформер равновесия - определяет конформацию с самой низкой энергией. Часто выполняется перед расчетом структуры с использованием квантово-химической модели.
- Распределение конформеров - получение набора низкоэнергетических конформеров. Обычно используется для определения формы, которую может принять конкретная молекула, и для определения распределения Больцмана для вычисления средних молекулярных свойств.
- Библиотека конформера - определяет конформер с наименьшей энергией и связывает его с набор конформеров, охватывающих все формы, доступные молекуле, независимо от энергии. Используется для создания библиотек для анализа подобия.
- Энергетический профиль - перемещает молекулу или систему через определенный пользователем набор координат, обеспечивая равновесную геометрию для каждого шага (с учетом ограничений, заданных пользователем).
- Сходство анализ - количественно оценивает сходство молекул (и, возможно, их конформеров) на основе структуры или химической функции (водородная связь акцепторы-доноры, положительные-отрицательные ионизируемые вещества, гидрофобные группы, ароматические углеводороды ). Определяет сходство молекулы (и, возможно, ее конформеров) с фармакофором.
Графическим пользовательским интерфейсом
Программное обеспечение содержит интегрированный графический пользовательский интерфейс. Операции с сенсорным экраном поддерживаются для устройств Windows 7 и 8. Построение молекул в 3D облегчается с помощью строителей молекул (включая органические, неорганические, пептидные, нуклеотидные и заместители). 2D-построение поддерживается для органических молекул с помощью палитры 2D-эскизов. Интерфейс версии Windows может обращаться к ChemDraw ; какие версии 9.0 или более поздние также могут использоваться для построения молекул в 2D. Диалог расчетов используется для уточнения задачи и метода расчета. Данные расчетов отображаются в диалогах или в виде текста. Дополнительный анализ данных, включая линейную регрессию, возможен из внутренней электронной таблицы.
Графические модели

Частичный вид карты электростатического потенциала
фуллерена (C60) синяя область внутри молекулы - это область положительного заряда (относительно надстройки, что дает наглядное объяснение способности фуллерена инкапсулировать отрицательно заряженные частицы).
Графические модели, особенно молекулярные орбитали, электронная плотность и электростатический потенциал карты, являются обычным средством молекулярной визуализации в химическом образовании.
- Поверхности:
- Молекулярные орбитали (самая высокая занятая, самая низкая незанятая и другие)
- Электронная плотность - Плотность, ρ ( r), является функцией координат r, определяемой таким образом, что ρ (r) dr - количество электронов внутри малого объема dr. Это то, что измеряется в эксперименте по дифракции рентгеновских лучей . Плотность может быть представлена в виде изоповерхности (поверхности изоплотности), при этом размер и форма поверхности задаются значением (или процентом отложения) электронной плотности.
- Спиновая плотность - Плотность, ρ (r) определяется как разница в электронной плотности, образованной электронами со спином α, ρα (r), и электронной плотностью, образованной электронами со спином β, ρβ (r). Для молекул с закрытой оболочкой (в которых все электроны спарены) спиновая плотность везде равна нулю. Для молекул с открытой оболочкой (в которых один или несколько электронов неспарены) спиновая плотность указывает на распределение неспаренных электронов. Плотность спина является показателем реакционной способности радикалов.
- Радиус Ван-дер-Ваальса (поверхность)
- Растворитель доступная площадь поверхности
- Электростатический потенциал - потенциал, ε p, определяется как энергия взаимодействия положительного точечного заряда, расположенного в точке p, с ядрами и электронами молекулы. Поверхность с отрицательным электростатическим потенциалом (поверхность с отрицательным потенциалом) очерчивает области молекулы, подверженные электрофильному воздействию.
- Составные поверхности (карты):
- Карта электростатического потенциала (электрофильный индикатор) - Наиболее часто используемой картой свойств является карта электростатического потенциала. Это дает потенциал в точках на определенной поверхности, чаще всего на поверхности с электронной плотностью, соответствующей общему размеру молекулы.
- Карта локального ионизационного потенциала - определяется как сумма орбитальных электронных плотностей, умноженная на ρi (r) абсолютные орбитальные энергии, ∈i, и деленные на полную плотность электронов, ρ (r). Потенциал локальной ионизации отражает относительную легкость удаления электронов («ионизации») в любом месте вокруг молекулы. Например, поверхность с «низким» потенциалом локальной ионизации для тетрафторида серы размечает области, которые наиболее легко ионизируются.
- Карта LUMO (нуклеофильный индикатор) - Карты молекулярных орбиталей также могут приводить к графическим индикаторам. Например, карта LUMO, на которой (абсолютное значение) самой низкой незанятой молекулярной орбитали (LUMO) отображается на размерную поверхность (опять же, чаще всего электронная плотность), обеспечивая указание нуклеофильная реакционная способность.
Спектральные расчеты

Рассчитанные (DFT / EDF2 / 6-31G *) ИК-спектры (красный), масштабированные и оптимизированные для экспериментальных FT-IR-спектров (синий) фенил-9-акридинкарбоксилата (ниже).

2D-рендеринг

3D-рендеринг Молекула фенил-9-акридинкарбоксилата.
Доступные спектральные данные и графики для:
Экспериментальные спектры могут быть импортированы для сравнения с рассчитанными спектрами: ИК- и УФ / видимые спектры в Объединенный комитет по атомным и молекулярно-физическим данным (JCAMP) (.dx) формат и ЯМР-спектры в формате Chemical Markup Language (.cml). Доступ к общедоступным спектральным базам данных доступен для спектров ИК, ЯМР и УФ / видимой области.
Базы данных
Spartan обращается к нескольким внешним базам данных.
- Базы данных квантово-химических расчетов:
- База данных Spartan Spectra Properties (SSPD) - набор из примерно 252 000 молекул со структурами, энергиями, ЯМР- и ИК-спектрами и волновыми функциями, рассчитанными с использованием EDF2 теория функционала плотности с базисным набором 6-31G * .
- Spartan Molecular Database (SMD) - набор примерно из 100 000 молекул, рассчитанных по следующим моделям:
- Hartree – Fock с 3- 21G, 6-31G * и 6-311 + G ** базисные наборы
- B3LYP функционал плотности с базисными наборами 6-31G * и 6-311 + G **
- EDF1 функционал плотности с базовым набором 6-31G *
- MP2 с базисом 6-31G * и 6-311 + G **
- G3 (MP2)
- T1
- Экспериментальные базы данных:
- NMRShiftDB - база данных с открытым исходным кодом экспериментальных химических сдвигов H и C.
- Cambridge Structural Database (CSD) - большой репозиторий малых молекул органических и неорганических экспериментальные кристаллические структуры около 600 000 записей.
- База данных NIST экспериментальных ИК- и УФ / видимых спектров.
История основных выпусков
- 1991 Spartan версия 1 Unix
- 1993 Spartan версия 2 Unix
- 1994 Mac Spartan Macintosh
- 1995 Spartan версия 3 Unix
- 1995 PC Spartan Windows
- 1996 Mac Spartan Plus Macintosh
- 1997 Spartan version 4 Unix
- 1997 PC Spartan Plus Windows
- 1999 Spartan version 5 Unix
- 1999 ПК Spartan Pro Windows
- 2000 Mac Spartan Pro Macintosh
- 2002 Spartan'02 Unix, Linux, Windows, Mac
Windows, Macintosh, версии Linux
- 2004 Spartan'04
- 2006 Spartan'06
- 2008 Spartan'08
- 2010 Spartan'10
- 2013 Spartan'14
- 2016 Spartan'16
- 2018 Spartan'18
См. Также
Ссылки
Внешние ссылки
- Официальный сайт, Wavefunction, Inc.

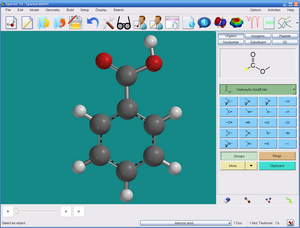 Графический интерфейс пользователя Spartan
Графический интерфейс пользователя Spartan Расчетная теплота образования T1 (ось y) относительно экспериментальной теплоты образования (ось x) для набора>1800 различных органических молекул из термохимической базы данных NIST со средним абсолютным и среднеквадратичным значением ошибки 8,5 и 11,5 кДж / моль соответственно.
Расчетная теплота образования T1 (ось y) относительно экспериментальной теплоты образования (ось x) для набора>1800 различных органических молекул из термохимической базы данных NIST со средним абсолютным и среднеквадратичным значением ошибки 8,5 и 11,5 кДж / моль соответственно.  Частичный вид карты электростатического потенциала фуллерена (C60) синяя область внутри молекулы - это область положительного заряда (относительно надстройки, что дает наглядное объяснение способности фуллерена инкапсулировать отрицательно заряженные частицы).
Частичный вид карты электростатического потенциала фуллерена (C60) синяя область внутри молекулы - это область положительного заряда (относительно надстройки, что дает наглядное объяснение способности фуллерена инкапсулировать отрицательно заряженные частицы).  Рассчитанные (DFT / EDF2 / 6-31G *) ИК-спектры (красный), масштабированные и оптимизированные для экспериментальных FT-IR-спектров (синий) фенил-9-акридинкарбоксилата (ниже).
Рассчитанные (DFT / EDF2 / 6-31G *) ИК-спектры (красный), масштабированные и оптимизированные для экспериментальных FT-IR-спектров (синий) фенил-9-акридинкарбоксилата (ниже).  2D-рендеринг
2D-рендеринг  3D-рендеринг Молекула фенил-9-акридинкарбоксилата.
3D-рендеринг Молекула фенил-9-акридинкарбоксилата.