Войти
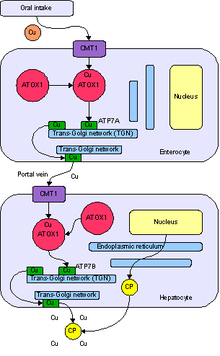 Физиологический путь меди в организме человека. Cu = медь, CP = церулоплазмин, белок ATP7B находится в гепатоците.
Физиологический путь меди в организме человека. Cu = медь, CP = церулоплазмин, белок ATP7B находится в гепатоците. Простая модель структурной особенности белка ATP7B. Cu = медь-связывающий мотив
Простая модель структурной особенности белка ATP7B. Cu = медь-связывающий мотив белок болезни Вильсона (WND ), также известный как белок ATP7B, представляет собой переносящую медь АТФазу P-типа который кодируется геном ATP7B. Белок ATP7B расположен в сети транс-Гольджи печени и мозга и уравновешивает уровень меди в организме, выводя избыток меди в желчь и плазму. Генетическое нарушение гена ATP7B может вызвать болезнь Вильсона, заболевание, при котором медь накапливается в тканях, что приводит к неврологическим или психиатрическим проблемам и заболеваниям печени..
Белок болезни Вильсона связан с ATP7B геном, примерно 80 Kb, расположенным на человека хромосома 13 и состоит из 21 экзона. МРНК, транскрибируемая геном ATP7B, имеет размер 7,5 КБ и кодирует белок из 1465 аминокислот.
Ген является участником транспорта катионов P-типа АТФазы семейство и кодирует белок с несколькими трансмембранными доменами, консенсусной последовательностью АТФазы , шарнирным доменом, сайтом фосфорилирования и по крайней мере двумя предполагаемыми медью -вяжущих сайтов. Этот белок действует как мономер, экспортируя медь из клеток, например, отток меди из печени в желчь. Были охарактеризованы альтернативные транскрипционные варианты сплайсинга, кодирующие разные изоформы с различными клеточными локализациями. Болезнь Вильсона вызывается различными мутациями. Одной из распространенных мутаций является мутация одной пары оснований, H1069Q.
Белок ATP7B представляет собой переносящую медь АТФазу P-типа, синтезируемую как мембранный белок 165 кДа в линии клеток гепатомы человека и которая на 57% гомологична белку, ассоциированному с болезнью Менкеса ATP7A.
ATP7B состоит из нескольких домены :
Мотив CPC ( Cys-Pro-Cys) в трансмембранном сегменте 6 характеризует белок как переносящий тяжелый металл АТФазу.
Связывающий мотив меди также демонстрирует высокое сродство к ионам других переходных металлов, таких как цинк Zn ( II), кадмий Cd (II), золото Au (III) и ртуть Hg (II). Однако медь способна снижать аффинность связывания цинка при низкой концентрации и резко увеличивает аффинность связывания меди с увеличением концентрации, чтобы гарантировать сильное связывание между мотивом и медью.
Как АТФазы P-типа, ATP7B подвергается ауто- фосфорилирование ключевого консервативного остатка аспарагиновой кислоты (D) в мотиве DKTGT. Связывание АТФ с белком инициирует реакцию, и медь связывается с трансмембранной областью. Затем происходит фосфорилирование по остатку аспарагиновой кислоты в мотиве DKTGT с высвобождением Cu. Затем дефосфорилирование остатка аспарагиновой кислоты восстанавливает белок, готовый к следующему транспорту.
Большая часть белка ATP7B находится в транс-Гольджи сеть (TGN) гепатоцитов, которая отличается от своего гомологичного белка ATP7A. Небольшое количество ATP7B находится в головном мозге. Как белок, транспортирующий медь, одна из основных функций - это доставка меди к медьзависимым ферментам в аппарате Гольджи (например, холоцерулоплазмин (CPN)).
человеческое тело печень играет важную роль в регуляции меди, включая удаление лишней меди. ATP7B участвует в физиологическом пути в процессе удаления меди двумя способами: секретируя медь в плазму и выделяя медь в желчь.
ATP7B получает медь от цитозольного белка антиоксиданта 1 шаперона меди (ATOX1). Этот белок нацелен на ATP7B непосредственно в печени, чтобы транспортировать медь. ATOX1 переносит медь из цитозоля в металл-связывающий домен ATP7B, который контролирует каталитическую активность ATP7B.
Несколько мутаций в ATOX1 могут блокировать пути меди и вызывать болезнь Вильсона.
ATP7B взаимодействует с глутаредоксином-1 (GLRX). Последующий транспорт стимулируется за счет уменьшения внутримолекулярных дисульфидных связей за счет катализа GLRX.
Болезнь Вильсона возникает, когда накопление меди внутри печени вызывает и клетки деструкция и проявляются симптомы заболевания печени. Затем потеря выведения меди с желчью приводит к увеличению концентрации меди в моче и вызывает проблемы с почками. Следовательно, симптомы болезни Вильсона могут быть различными, включая заболевание почек и неврологическое заболевание. Основная причина - нарушение функции ATP7B из-за мутаций одной пары оснований, делеций, сдвигов рамки считывания, ошибок сплайсинга в гене ATP7B.