
Войти
| Синдром Ваарденбурга | |
|---|---|
| Другие названия | Синдром Клейна – Ваарденбурга (тип 3), синдром Шаха – Ваарденбурга (тип 4) |
 | |
| Черты лица синдрома Ваарденбурга типа 1 (из описания Яна ван дер Хуве, 1916 г.) | |
| Специальность | Медицина генетика |
синдром Ваарденбурга - это группа редких генетических состояний, характеризующихся, по крайней мере, некоторой степенью врожденной потери слуха и дефектами пигментации, которые могут включать ярко-голубые глаза (или один синий глаз и один карий глаз ), белый чуб или участки светлой кожи. Эти основные характеристики составляют 2-й тип состояния; у типа 1 также имеется более широкая щель между внутренними уголками глаз, называемая telecanthus или dystopia canthorum. При типе 3, который встречается редко, руки и кисти также имеют неправильную форму с постоянными контрактурами пальцев или сросшимися пальцами, в то время как при типе 4 человек также имеет болезнь Гиршпрунга, что является врожденным недостатком нервов в кишечнике, приводящим к дисфункции кишечника. Также существует как минимум два типа (2E и PCWH), которые могут вызывать симптомы центральной нервной системы, такие как задержка развития и нарушения мышечного тонуса.
Синдром вызванные мутациями в любом из нескольких генов, которые влияют на деление и миграцию клеток нервного гребня во время эмбрионального развития (хотя некоторые из вовлеченные гены также влияют на нервную трубку ). Клетки нервного гребня - это стволовые клетки, оставшиеся после закрытия нервной трубки, которые продолжают формировать различные клетки не центральной нервной системы в разных частях тела, включая меланоциты, различные кости и хрящи лица и внутреннего уха и периферические нервы кишечника. Тип 1 вызван мутацией в гене PAX3, тогда как ген, который наиболее часто вызывает тип 2 при мутации, - это MITF. Тип 3 является более серьезным проявлением типа 1 и вызван мутацией в том же гене, тогда как тип 4 чаще всего вызывается мутацией в SOX10. Мутации в других генах также могут вызывать различные типы, и некоторым из них были присвоены собственные подтипы, обозначенные буквами. Большинство типов аутосомно-доминантное.
Предполагаемая распространенность синдрома Ваарденбурга составляет 1 на 42 000. Типы 1 и 2 являются наиболее распространенными, составляя примерно половину и треть случаев соответственно, в то время как тип 4 составляет пятый, а тип 3 - менее 2% случаев. По оценкам, 2–5% врожденно глухих людей страдают синдромом Ваарденбурга. Описание синдрома относится, по крайней мере, к первой половине 20 века, однако он назван в честь голландского офтальмолога и генетика Петруса Йоханнеса Ваарденбурга, который описал его в 1951 году. Его подтипы постепенно обнаруживались в последующем. десятилетия, и им приписывали гены в основном в 1990-х и 2000-х годах.
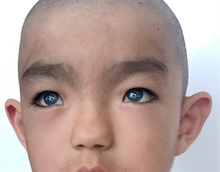 Внешний вид мальчика из Китая с синдромом Ваарденбурга 1 типа
Внешний вид мальчика из Китая с синдромом Ваарденбурга 1 типа Синдром Ваарденбурга имеет несколько разных типов с некоторыми вариациями симптомов, и симптомы могут различаться у людей одного типа. Двумя характеристиками, характерными для всех типов синдрома Ваарденбурга, являются некоторая степень врожденной нейросенсорной тугоухости и некоторая степень дефицита пигментации, наиболее характерная для глаз.
Тип 1 характеризуется врожденной сенсоневральной тугоухостью, пигментными недостатками волос, такими как белая прядь волос (полиоз ) в передней центральной части головы или преждевременное поседение, пигментный дефицит глаз, такой как глаза разного цвета (полная гетерохромия радужной оболочки ), несколько цветов в глазу (секторальная гетерохромия радужной оболочки) или ярко-голубые глаза, участки депигментации кожи и более широкий промежуток между внутренними углами глаза назывались telecanthus или dystopia canthorum. Другие черты лица, связанные с типом 1, могут включать высокую переносицу, плоский кончик носа, монобровь (синофрис), меньшие края ноздрей (крылья) или гладкий желобок.
Различие, которое отличает тип 2 от типа 1, заключается в том, что пациенты не имеют более широкого промежутка между внутренними уголками глаз (telecanthus / dystopia canthorum). Сенсорно-невральная тугоухость, как правило, встречается чаще и тяжелее при этом типе. Безусловно, наиболее распространенным геном, вызывающим этот тип при мутации, является MITF (классифицируется как тип 2A). Если у двух человек с мутацией в этом гене есть ребенок, несущий обе мутации (гомозиготный ), вероятность которого составляет 25%, у ребенка присутствуют дополнительные симптомы, такие как отверстие в радужной оболочке (колобома ), маленькие глаза (микрофтальм ), затвердевшие кости (остеопетроз ), макроцефалия, альбинизм и глухота.
Известны два пациента с мутациями в обеих копиях SNAI2 (классифицируется как тип 2D); эти люди имели синдром Ваарденбурга типа 2, но не имели дефектов пигментации волос.
Когда синдром Ваарденбурга типа 2 вызван мутацией в SOX10 (классифицируется как тип 2E), он может в некоторых случаях проявляются множественные неврологические симптомы. Они могут включать задержку развития, нистагм в раннем детстве, повышенный мышечный тонус, аномалии белого вещества или гипомиелинизацию в головном мозге, аутистическое -подобное поведение и недоразвитие или полное отсутствие многих структур внутреннего уха, таких как вестибулярная система или улитка. Также может присутствовать отсутствие обоняния (аносмия ) из-за отсутствия обонятельной луковицы в головном мозге.
Также известный как синдром Клейна-Ваарденбурга или синдром Ваарденбурга-Клейна, тип 3 имеет те же симптомы, что и тип 1 (и вызывается мутациями в том же гене), но имеет дополнительные симптомы, которые влияют на руки и кисти. Это могут быть суставные контрактуры пальцев (камптодактилия ), вызванные недоразвитостью мышц, а также сращение пальцев (синдактилия ) или крылатые лопатки. Также возможны микроцефалия и задержка развития.
Также известный как синдром Шаха-Ваарденбурга или синдром Ваарденбурга-Шаха, тип 4 имеет большинство тех же характеристик как тип 2 (то есть без телекантуса или очевидной более широкой глазной щели), но с добавлением болезни Гиршпрунга, которая представляет собой врожденное отсутствие нервов в кишечнике, ведущее к дисфункции кишечника. Кроме того, потеря слуха встречается не так часто, как при типе 2. Редко заячья губа сообщается при этой форме синдрома Ваарденбурга.
Тип 4 также может быть вызван мутацией в SOX10 (тот же ген, что и у типа 2E), в котором он известен как тип 4C; Потеря слуха у этого типа очень распространена и серьезна.
Мутация в SOX10, гене, участвующем в типах 2E и 4C, иногда может приводить к симптомам обоих типов (неврологические симптомы, которые иногда наблюдаются при типе 2E, и болезни Гиршпрунга, как при типе 4). Когда это происходит, это называется периферической демиелинизирующей нейропатией - центральной дисмиелинизирующей лейкодистрофией - синдромом Ваарденбурга - болезнью Гиршпрунга (PCWH).
 Синдром Ваарденбурга обычно наследуется по аутосомно-доминантному типу.
Синдром Ваарденбурга обычно наследуется по аутосомно-доминантному типу.  Типы 2D, 3, 4A и 4B иногда могут иметь аутосомно-рецессивный тип наследования.
Типы 2D, 3, 4A и 4B иногда могут иметь аутосомно-рецессивный тип наследования. Синдром Ваарденбурга вызывается мутациями в любом из нескольких генов, влияющих на операцию клеток нервного гребня в эмбриональном развитии. Большинство типов синдрома Ваарденбурга вызываются аутосомно-доминантными мутациями. Аутосомно-рецессивные редки. В большинстве случаев больной унаследовал его от одного из родителей с одной из доминирующих форм заболевания. Небольшой процент случаев является результатом спонтанных новых мутаций в гене, при отсутствии семейной истории болезни.
Нервный гребешок представляет собой группу временных мигрирующих клеток, которые остаются после закрытия нервной трубки (нейруляция ), примерно на четвертой неделе эмбрионального развития. Они несут ответственность за дифференциацию в разнообразную группу клеток, которые достигают разных областей тела. Нервная трубка и нервный гребень происходят из эктодермы ; нервная трубка продолжает формировать головной и спинной мозг, тогда как клетки нервного гребня в конечном итоге продолжают формировать различные кости и хрящи черепа и лица, мигрируя через глоточные дуги. Они также дифференцируются в stria vascularis улитки, нервы и глию кишечника (миэнтерическое сплетение ), Шванновские клетки, которые миелинизируют периферическую нервную систему для обеспечения достаточной проводимости, одонтобласты, которые продуцируют дентин глубоко в зубах, некоторые нейроэндокринные клетки, соединительная ткань вокруг слюнной, слезной, гипофиза, тимуса и щитовидная железа, соединительная ткань глаза, такая как строма радужной оболочки и роговица и трабекулярная сеть, и меланоциты, включая те, которые находятся в строме радужной оболочки, которые вызывают коричневый цвет глаз через меланин. Клетки нервного гребня также играют роль в формировании мышц, в том числе стеночных мышц определенных сердечных артерий.
Исследование было проведено на редком случае двойного гетерозиготного ребенка, когда каждый родитель имел только одиночные мутации в MITF или PAX3. Эффект двойных гетерозиготных мутаций в генах MITF и PAX3 в WS1 и WS2 может усиливать симптомы поражения пигмента. Это приводит к выводу, что двойная мутация MITF связана с конечностью синдрома Ваарденбурга и может влиять на фенотипы или симптомы синдрома.
| Тип | OMIM | Ген | Локус | Наследование |
|---|---|---|---|---|
| Тип 1 (WS1) | 193500 | PAX3 | 2q36.1 | Аутосомно-доминантный |
| Тип 2A (WS2A, первоначально WS2) | 193510 | MITF | 3p14.1 – p12.3 | Аутосомно-доминантный |
| Тип 2B (WS2B) | 600193 | WS2B | 1p21 – p13.3 | Аутосомно-доминантный |
| Тип 2C (WS2C) | 606662 | WS2C | 8p23 | Аутосомно-доминантный |
| Тип 2D (WS2D) | 608890 | SNAI2 | 8q11 | Аутосомно-рецессивный |
| Тип 2E (WS2E) | 611584 | SOX10 | 22q13.1 | Аутосомно-доминантный |
| Тип 3 (WS3) | 148820 | PAX3 | 2q36.1 | Аутосомно-доминантный или аутосомно-рецессивный |
| Тип 4A (WS4A) | 277580 | EDNRB | 13q22 | Аутосомно-доминантный или аутосомно-рецессивный |
| Тип 4B (WS4B) | 613265 | EDN 3 | 20q13 | Аутосомно-доминантный или аутосомно-рецессивный |
| Тип 4C (WS4C) | 613266 | SOX10 | 22q13.1 | Аутосомно-доминантный |
В настоящее время нет лечения или лекарства от синдрома Ваарденбурга. Симптомом, который, скорее всего, имеет практическое значение, является глухота, и к ней относятся так же, как к любой другой необратимой глухоте. В отмеченных случаях могут возникнуть косметические проблемы. Другие аномалии (неврологические, структурные, болезнь Гиршпрунга ), связанные с синдромом, лечат симптоматически.
Распространенность всех типов синдрома Ваарденбурга оценивается примерно в 1 случай на 42 000. Типы 1 и 2 являются наиболее распространенными, а тип 1 - немного более распространенным. В обзоре, проведенном в 2015 году с участием 417 пациентов, было обнаружено, что тип 1 является наиболее распространенным типом, охватывая около половины всех случаев (47%), в то время как тип 2 был вторым по распространенности типом, охватывая около трети (33%).. Подавляющее большинство (около 85%) случаев 2-го типа относятся к типу 2А. Распространенность типа 2B неизвестна, поскольку о нем сообщалось только в одном исследовании 1996 года. Тип 2C до сих пор был обнаружен только в одной итальянской семье, а тип 2D был обнаружен только у 2 неродственных пациентов по состоянию на 2018 год. Количество известных случаев типа 2E, связанных с неврологическими аномалиями, по состоянию на 2017 год составляло 23, в то время как количество остальных неизвестно. Тип 3 встречается реже, чем типы 1, 2 и 4, и составляет менее 2% случаев. Тип 4, по-видимому, составляет около пятой части случаев (19%). Из его подтипов тип 4C является наиболее распространенным (около 71% от типа 4), за ним следуют тип 4A (19%) и тип 4B (10%).
Считается, что синдром Ваарденбурга является наиболее распространенным. присутствует у 2–5% врожденно глухих. Врожденная глухота составляет около половины глухоты в целом. Примерно 1 из 30 учеников школ для глухих страдает синдромом Ваарденбурга. Различное проявление синдрома затрудняет получение точных цифр его распространенности.
В 1916 году голландский офтальмолог Ян ван дер Хов (1878 г.) –1952) описал пару девочек-близнецов с глухотой и особым типом блефарофимоза, который считается dystopia canthorum, обнаруживаемым при синдроме Ваарденбурга типов 1 и 3. Блефарофимоз описывает веки, которые недоразвиты так, что они постоянно покрывают часть глаз.
В 1926 году немецкий врач Ирмгард Менде описал семью из четырех поколений, в которой пятеро детей имели симптомы депигментации волос, кожи и глаз, глухоту и "монголоидную " внешность. (Позже Ваарденбург приписал это описание dystopia canthorum.) Это позже привело к тому, что синоним синдрома Менде был зарегистрирован в некоторых базах данных.
В 1929 году голландский врач К.Т.А. Хальбертсма описал семейную модель dystopia canthorum, а в 1930 году Итальянский врач Винченцо Гуальди (1891–1976) также подтвердил наследственную картину dystopia canthorum. Позднее это привело к тому, что в некоторых базах данных был зарегистрирован синоним Ван дер Хов-Хальбертсма-Ваарденбург-Гуальди.
В 1947 году швейцарский офтамолог Дэвид Кляйн (1908–1993) впервые сообщил о пациенте при двусторонней глухоте, нарушениях пигментации, характерных чертах лица и пороках развития рук. Хотя это было первое полное описание пациента с синдромом Ваарденбурга 3-го типа, современные клиницисты не считают описанный им синдром таким же, как описанный Ваарденбургом четыре года спустя, отчасти из-за того, насколько серьезными были пороки развития руки в его теле.
Синдром был впервые полностью формализован и описан голландским офтальмологом и генетиком Петрусом Йоханнесом Ваарденбургом (1886–1979) в 1951 году. Состояние, которое он описал, теперь классифицируется как синдром Ваарденбурга типа 1.
Тип 2 был впервые установлен в 1971 году, когда в ходе исследования было обнаружено, что у некоторых пациентов с синдромом Ваарденбурга не было канторной дистопии. Исследование 1977 года подтвердило семейный образец этого другого представления. Два исследования 1994 года впервые подтвердили связь между этим типом синдрома Ваарденбурга и мутациями в гене MITF (теперь классифицируемом как тип 2A), расположенном на хромосоме 3 в локусе 3p14.1 – p12.3.
Тип 2B был впервые установлен в 1994 году, когда в том же исследовании, в котором были обнаружены мутации в MITF у пациентов с синдромом Ваарденбурга типа 2, также было обнаружено, что у некоторых пациентов не было никаких мутаций в этой области. Второе исследование 1994 г. обнаружило связь с хромосомой 1 в локусе 1p21 – p13.3. Это состояние стало известно как тип 2B (с геном, обозначенным WS2B ), однако с тех пор это не было зарегистрировано, и ответственный ген остается неизвестным.
Тип 2C был установлен в 2001 году. когда исследование итальянской семьи с признаками синдрома Ваарденбурга типа 2 показало, что они были вызваны неизвестным геном на хромосоме 8 в локусе 8q23, который был нарушен в результате хромосомной транслокации. В ходе исследования было установлено предварительное название гена: WS2C. Однако с тех пор мутации в этой области у пациентов с синдромом Ваарденбурга не обнаруживались.
Тип 2D был установлен в 2002 году, когда исследование, направленное на поиск мутаций в человеческой версии гена SNAI2, известно, что вызывает депигментацию у мышей, были обнаружены делеции обеих копий этого гена у 2 неродственных индивидуумов с синдромом Ваарденбурга типа 2. С тех пор мутации в обеих копиях этого гена не были обнаружены у людей с синдромом Ваарденбурга 2 типа.
Тип 2E был впервые установлен в 1996 году, когда исследование выявило девушку с симптомами синдрома Ваарденбурга типа 2, но с дополнительным недоразвитием передней части глаза, что привело к слепоте. В 1999 году было обнаружено, что у нее была мутация в гене SOX10, а более поздние исследования подтвердили связь между мутациями в этом гене и этим фенотипом, а также неврологические симптомы, такие как задержка развития.
Тип 3 впервые получил свое название от Goodman et al. в 1981 году в сотрудничестве с Klein, в котором они установили связь с аномалиями рук, о которых впервые сообщил Кляйн в 1947 году. Мутации в PAX3 были впервые связаны с этим фенотипом в 1992 году.
Коморбидность с болезнью Гиршпрунга, который позже будет составлять тип 4, впервые был замечен в различных исследованиях в 1970-х годах. Индийский педиатр Кришнакумар Шах и его коллеги впервые обозначили этот синдром как возможный вариант синдрома Ваарденбурга в 1981 году. Впервые этот вариант был приписан мутации в EDNRB в 1994 году (теперь классифицируется как тип 4A). Тип 4B был установлен в 1996 году, когда было обнаружено, что мутации в EDN3 приводят к этому типу синдрома Ваарденбурга, а тип 4C был впервые установлен в 1998 году, когда также было обнаружено, что мутации в SOX10 приводят к к этому типу.
 Глухая белая кошка с гетерохромией
Глухая белая кошка с гетерохромией синдром Ваарденбурга типа 2A (с мутацией в MITF) обнаружен у собак, Крупный рогатый скот Fleckvieh, норки, мыши и золотой хомяк. Дегенерация улитки и мешочка, наблюдаемая при синдроме Ваарденбурга, также была обнаружена у глухих белых кошек, далматинов и других пород собак, белых норок и мышей.
Домашние кошки. с голубыми глазами и белыми халатами часто бывают полностью глухими. Глухота гораздо чаще встречается у белых кошек, чем у кошек других окрасов. Согласно ASPCA Complete Guide to Cats, «от 17 до 20 процентов белых кошек с не голубыми глазами глухие; 40 процентов« разноглазых »белых кошек с одним голубым глазом глухие; и 65 до 85 процентов голубоглазых белых кошек глухие ». Хотя было проведено мало исследований, чтобы связать это с генами, которые, как известно, участвуют в синдроме Ваарденбурга у человека, генетическое нарушение развития нервного гребня может привести к этому проявлению и у кошек. Было обнаружено, что один из генов, который приводит к глухоте и белой шерсти у кошек при мутации, KIT, увеличивает экспрессию MITF.
Летальный белый синдром - это синдром у лошадей, вызванный мутациями. в обоих экземплярах ЕДНРБ. Это приводит к смерти от псевдообструкции кишечника из-за болезни Гиршпрунга. Однако мутация в единственной копии EDNRB, как и при синдроме Ваарденбурга типа 4A, приводит к образованию пятнистой белой шерсти оверо с глухотой.
Хорьки с синдромом Ваарденбурга имеют небольшую белую полосу вдоль на макушке или затылке, а иногда и по задней части шеи (известный как «пламенеющий» рисунок шерсти), или сплошная белая голова от носа до плеч (известная как рисунок шерсти «панды»). У пораженных хорьков череп часто немного более плоский, а глаза широко посажены, чем у здоровых хорьков. Поскольку у здоровых хорьков плохой слух, глухоту можно определить только по отсутствию реакции на громкие звуки. Поскольку это наследственное заболевание, пораженных животных нельзя использовать для разведения. Изучение корреляции между вариациями шерсти и глухотой у европейских хорьков показало: «Все (n = 27) панда, американская панда и огненные хорьки были глухими».
.
| Классификация | D |
|---|---|
| Внешние ресурсы |
.