
Войти
| Иминоглицинурия | |
|---|---|
| Другие названия | Семейная иминоглицинурия |
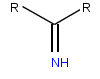 | |
| Имин, функциональная группа, обнаруженная в иминокислотах | |
| Специальность | Эндокринология |
Иминоглицинурия, представляет собой аутосомное рецессивное нарушение почечного канальцевого транспорта, влияющее на реабсорбцию аминокислоты глицина, и иминовые кислоты пролин и гидроксипролин. Это приводит к избыточному моче экскреции всех трех кислот (-урия означает «в моче»).
Иминоглицинурия - редкое и сложное заболевание, связанное с количество генетических мутаций, которые вызывают дефекты как в почечной, так и в кишечной транспортных системах глицина и иминокислот.
Иминокислоты обычно содержат имин функциональная группа вместо аминогруппы, обнаруженной в аминокислотах. Пролин считается и обычно называют аминокислотой, но, в отличие от других, он имеет вторичный амин. Эта особенность, уникальная для пролина, позволяет идентифицировать пролин также как иминокислоту. Гидроксипролин - еще одна иминокислота, полученная в результате естественного гидроксилирования пролина.
Основной характеристикой иминоглицинурии является присутствие глицина и иминокислот в моче. В противном случае это считается относительно доброкачественным заболеванием, хотя симптомы, связанные с нарушениями метаболизма пролина и глицина, вызванными мальабсорбцией, могут присутствовать при иминоглицинурии. К ним относятся энцефалопатия, умственная отсталость, глухота, слепота, камни в почках, гипертония и круговая атрофия.
Циркулярная атрофия - это наследственное дегенеративное заболевание сетчатки и сосудистой оболочки, иногда сопровождающее метаболическое состояние гиперорнитинемия. Наличие спиральной атрофии с иминоглицинурией происходит из-за дефицита пролина в хориоретинальных тканях, в то время как процессы, лежащие в основе гиперорнитинемии, нарушают метаболический путь от орнитина к пролину, что изменяет катаболизм орнитина, а также приводит к снижению уровня пролина. Таким образом, спиральная атрофия может быть обнаружена при любом заболевании с недостаточностью пролина в качестве основного признака.
Гиперглицинурия - еще одно заболевание, влияющее на реабсорбцию глицина и иминокислот, подобное иминоглицинурии и считается гетерозиготным форма. Когда он сопровождается определенным типом почечного камня (нефролитиаз), его иногда называют «иминоглицинурией, тип II».
 Иминоглицинурия имеет аутосомно-рецессивный тип наследования.
Иминоглицинурия имеет аутосомно-рецессивный тип наследования.Считается, что иминоглицинурия передается по аутосомно-рецессивному типу. Это означает, что дефектный ген, ответственный за заболевание, расположен на аутосоме, а для наследования требуется две копии дефектного гена - по одной от каждого родителя. Оба родителя человека с аутосомно-рецессивным заболеванием несут одну копию дефектного гена, но обычно не испытывают никаких признаков или симптомов заболевания.
Ненаследственная причина избытка Экскреция пролина и глицина с мочой, как и при иминоглицинурии, довольно часто встречается у новорожденных младше 6 месяцев. Иногда это называют неонатальной иминоглицинурией, это связано с неразвитостью высокоаффинных транспортных механизмов в почечном контуре, в частности, PAT2, SIT1 и SLC6A18. Состояние исправляется с возрастом. Однако в случаях, когда это сохраняется после детства, можно заподозрить наследственную гиперглицинурию или иминоглицинурию.
Глицин, пролин и гидроксипролин имеют общие почечные канальцевые механизмы реабсорбции, функция, специфичная для проксимального канальца. Как реабсорбция, так и абсорбция глицина и иминокислот происходит соответственно в проксимальных канальцах или кишечном щеточном кайме эпителии. Более избирательный транспорт пролина и других иминокислот управляется на молекулярном уровне механизмом клеточного транспорта млекопитающих, удачно известным как система IMINO.
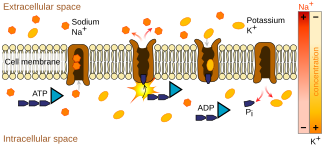 Активный транспорт в клетку через ионные каналы с использованием способности связывания обеспечивается обменом натрия и калия.
Активный транспорт в клетку через ионные каналы с использованием способности связывания обеспечивается обменом натрия и калия. Хотя ни одна генетическая мутация не была установлена как причина иминоглицинурии; Известно, что с нарушением связаны несколько мутаций, влияющих на транспортные механизмы, общие для глицина, пролина и гидроксипролина, а также мутации, которые избирательно переносят глицин или иминокислоты, включая систему IMINO. В сочетании эти факторы приведут к вариабельному фенотипу для иминоглицинурии в зависимости от присутствующих мутаций. Однако, несмотря на роль, которую кишечная мальабсорбция глицина и иминокислот может играть в иминоглицинурии, первичный дефект нарушает их почечный транспорт и реабсорбцию. Это очевидно, поскольку наследственная иминоглицинурия может клинически проявляться без вовлечения кишечника.
У млекопитающих, включая людей, транспорт аминокислот и иминокислот из просвета (внутренняя часть) кишечника или проксимального канальца почек в клетки происходит на мембране щеточной каймы эпителия (влажная, плотно упакованная клеточная выстилка многих тканей и органов тела). Здесь котранспортеры, такие как натрия или хлорид (часть системы котранспортеров Na-K-Cl ), соединяются с амино или иминокислоты на молекулярном уровне и транспортируют их через специфические интегральные мембранные белки, которые образуют ионные каналы, которые расположены внутри клеточной мембраны. Из клеток абсорбированные или реабсорбированные аминокислоты и иминокислоты в конечном итоге достигают крови. Абсорбция относится к общему процессу, происходящему в кишечнике вместо нормального пищеварительного расщепления белков, в то время как реабсорбция относится к процессу, происходящему в проксимальных канальцах почек, с целью регенерации аминокислот и иминокислот, которые были отфильтрованы из крови через glomerulus.
Эти формы транспорта требуют энергии, поскольку транспортируемые продукты обычно движутся против более высокого градиента концентрации. Этот процесс, называемый активный транспорт, получает свою энергию от АТФ и других связанных с АТФ систем транспорта, которые производят энергию, таких как натриево-калиевый насос..
Первичный дефект, связанный с иминоглицинурией, представляет собой гомозиготную (рецессивную) мутацию гена SLC36A2 (PAT2). Один из нескольких мембранных транспортных белков в семействе переносчиков растворенных веществ переносчиков аминокислот, PAT2 является высокоаффинным почечным переносчиком глицина, пролина и гидроксипролина, который, как обнаружено, является дефектным в обоих аллели при наличии иминоглицинурии у человека. Это контрастирует с тем фактом, что, когда дефектен только один аллель PAT2, вместо иминоглицинурии будет присутствовать гиперглицинурия. Эти данные определяют иминоглицинурию как гомозиготную форму гиперглицинурии, причем первая имеет более высокую степень экскреции с мочой глицина и иминокислот, коррелирующих с мутациями в обоих аллелях.
Может быть обнаружена еще одна мутация, предположительно передающая фенотип иминоглицинурии. в гене SLC36A1 (PAT1). PAT1, идентифицированный как кишечный переносчик глицина и иминокислот с низким сродством, работает в сотрудничестве с почечным натрий-водородным обменником NHE3 (SLC9A3 ). Поскольку абсорбция и реабсорбция глицина, пролина и гидроксипролина также происходит через PAT1, считается, что он играет еще одну роль в проявлении фенотипа мальабсорбтивной иминоглицинурии. Однако в недавних сообщениях предполагается, что роль PAT1 в некоторых случаях заболевания снижается.
Хотя PAT2 явно указывается как первичный мутаген, ответственный за иминоглицинурию, вариабельность фенотипа определяется тремя изменение генетических мутаций. Основным из них считается система IMINO.
Определяется как натрийзависимый переносчик пролина, не ингибируемый аланином, система IMINO, которая, как полагают, образуется SLC6A20 Ген (SIT1) является важным транспортным механизмом млекопитающих, ответственным как за реабсорбцию почек, так и за абсорбцию в кишечнике пролина и других иминокислот, таких как гидроксипролин и. Последовательность мРНК для SIT1 экспрессируется в большей части желудочно-кишечного тракта, включая желудок, двенадцатиперстную кишку, тощую кишку., подвздошная кишка, слепая кишка и толстая кишка. Он также обнаружен в почках, оптической сосудистой оболочке и частях центральной нервной системы, таких как мозг и микроглиальные клетки. 157>
Пониженная пенетрантность - это явление, при котором полностью унаследованный генетический признак, такой как заболевание или расстройство, не может проявлять ожидаемый фенотип. Об этом сообщалось в некоторых случаях иминоглицинурии. Здесь предполагается, что система IMINO играет роль в снижении пенетрантности иминоглицинурии, компенсируя мальабсорбцию иминокислоты, специфически связанную с мутациями PAT2. И наоборот, считается, что мутации SIT1 приводят к полной экспрессии иминоглицинурии в некоторых случаях, когда гетерозиготные мутации PAT2 в противном случае были бы достаточны только для того, чтобы вызвать гиперглицинурию.
Считается, что две другие транспортные системы играют последующие роли в иминоглицинурии, когда в них присутствуют мутации. Переносчик нейтральных аминокислот SLC6A19 (влияющий на глицин, пролин и другие нейтральные аминокислоты, такие как цистеин и триптофан ), связанный с болезнью Хартнупа, играет роль в иминоглицинурии как модификатор мутаций PAT2, а также напрямую зависит от действия SIT1. Глицин-специфический переносчик, SLC6A18, также влияет на фенотип иминоглицинурии, либо усложняя, либо компенсируя нарушения транспорта глицина.
Подводя итог, иминоглицинурия в первую очередь выражается гомозиготными мутациями почечный транспортер PAT2, тогда как общий фенотип иминоглицинурии может быть изменен нормальной или дефектной активностью SIT1 (IMINO), SLC6A19 и SLC6A18.
| Классификация | D |
|---|---|
| Внешние ресурсы |