
Войти
| Синдром Тричера Коллинза | |
|---|---|
| Другие названия | Синдром Тричера Коллинза – Франческетти, нижнечелюстной дизостоз, Франческетти-Цвален -Синдром Клейна |
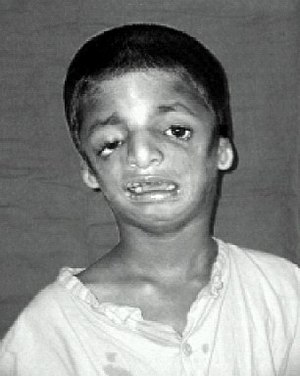 | |
| Ребенок с синдромом Тричера-Коллинза | |
| Специальность | Медицинская генетика |
| Симптомы | Деформации ушей, глаз, скул, подбородка |
| Осложнения | Проблемы с дыханием, проблемы со зрением, потеря слуха |
| Причины | Генетические |
| Метод диагностики | На основании симптомов, Рентген, генетическое тестирование |
| Дифференциальный диагноз | синдром Нагера, синдром Миллера, гемифациальная микросомия |
| Лечение | Реконструктивная хирургия, слуховые аппараты, логопед |
| Прогноз | В целом нормальный ожидаемая продолжительность жизни |
| Частота | 1 из 50 000 человек |
Синдром Тричера Коллинза (TCS ) - это генетическое заболевание, характеризующееся деформациями ушей, глаз, скул и подбородка.. Однако степень поражения человека может варьироваться от легкой до тяжелой. Осложнения могут включать проблемы с дыханием, проблемы со зрением, волчью пасть и потерю слуха. Пострадавшие обычно имеют средний интеллект.
TCS обычно аутосомно-доминантный. Более чем в половине случаев это происходит в результате новой мутации , а не по наследству от родителей человека. Участвующие гены могут включать TCOF1, POLR1C или POLR1D. Диагноз обычно подозревается на основании симптомов и рентгеновских лучей, и потенциальное подтверждение генетическим тестированием.
синдром Тричера Коллинза неизлечим. Симптомы можно лечить с помощью реконструктивной хирургии, слуховых аппаратов, логопеда и других вспомогательных устройств. Продолжительность жизни в целом нормальная. TCS встречается примерно у одного из 50 000 человек. Синдром назван в честь Эдварда Тричера Коллинза, английского хирурга и офтальмолога, которые описали его основные черты в 1900 году.
 Тот же ребенок, показанный спереди вверху в информационном окне, теперь виден сбоку, с маленькими ушами и подбородком, который находится далеко позади.
Тот же ребенок, показанный спереди вверху в информационном окне, теперь виден сбоку, с маленькими ушами и подбородком, который находится далеко позади. Симптомы у людей с синдромом Тричера Коллинза различаются. У некоторых людей поражение настолько легкое, что их диагноз остается невыявленным, в то время как у других наблюдается умеренное или тяжелое поражение лица и опасные для жизни нарушения дыхательных путей. Большинство признаков ТКС симметричны и распознаются уже при рождении.
Наиболее частым симптомом синдрома Тричера-Коллинза является недоразвитие нижней челюсти и скуловой кости.. Это может сопровождаться втягиванием язычка. Маленькая нижняя челюсть может привести к плохой окклюзии зубов или, в более тяжелых случаях, затрудненному дыханию или глотанию. Дыхательная система ребенка с синдромом Тричера Коллинза является основной проблемой, когда ребенок рождается, и другие проблемы решаются после того, как будут решены респираторные проблемы. Недоразвитие скуловой кости придает щекам запавший вид.
Наружное ухо иногда маленькое, повернуто, деформировано или полностью отсутствует у людей с ТКШ. Также описывается симметричное двустороннее сужение или отсутствие наружного слухового прохода. В большинстве случаев кости среднего уха и полость среднего уха деформированы. Пороки развития внутреннего уха описываются редко. В результате этих аномалий у большинства людей с TCS наблюдается кондуктивная потеря слуха.
У большинства людей также возникают проблемы со зрением, в том числе колобомы (выемки) в нижних веках, частичные или частичные. полное отсутствие ресниц на нижнем веке, веки, наклоненные вниз, опущение верхнего и нижнего века и сужение слезных протоков. Может возникнуть потеря зрения, связанная с косоглазием, аномалиями рефракции и анизометропией. Это также может быть вызвано сильной сухостью глаз, следствием аномалий нижних век и частых глазных инфекций.
Хотя череп неправильной формы не характерен для синдрома Тричера Коллинза, брахицефалия с битемпоральным сужением иногда наблюдается. Расщелина неба также является обычным явлением.
Стоматологические аномалии наблюдаются у 60% пострадавших людей, включая агенез зубов (33%), изменение цвета (помутнение эмали) (20%), неправильное смещение зуба. первые моляры верхней челюсти (13%) и большое расстояние между зубами. В некоторых случаях аномалии зубов в сочетании с гипоплазией нижней челюсти приводят к неправильному прикусу. Это может привести к проблемам с приемом пищи и невозможностью закрыть рот.
Менее распространенные признаки TCS могут усугубить проблемы с дыханием у пострадавшего человека, включая апноэ во сне. Атрезия хоан или стеноз - это сужение или отсутствие хоан, внутреннего отверстия носовых ходов, которое также может наблюдаться. Недоразвитие глотки также может сужать дыхательные пути.
Реже встречаются признаки, связанные с TCS, включая деформации носа, высокое арочное небо, макростомию, преаурикулярные волосы смещение, волчья пасть, гипертелоризм, выемка на верхнем веке и врожденные пороки сердца.
Хотя деформация лица часто связана с задержкой развития и умственной отсталостью, более 95% людей, страдающих TCS, имеют нормальный интеллект. Психологические и социальные проблемы, связанные с лицевой деформацией, могут повлиять на качество жизни людей с СКС.
 Синдром Тричера Коллинза наследуется по аутосомно-доминантной модели.
Синдром Тричера Коллинза наследуется по аутосомно-доминантной модели. Мутации в генах TCOF1, POLR1C или POLR1D могут вызывать синдром Тричера Коллинза. Мутации гена TCOF1 являются наиболее частой причиной заболевания, а мутации генов POLR1C и POLR1D вызывают еще 2% случаев. У людей без идентифицированной мутации в одном из этих генов генетическая причина состояния неизвестна. Гены TCOF1, POLR1C и POLR1D кодируют белки, которые играют важную роль в раннем развитии костей и других тканей лица. Мутации в этих генах снижают производство рРНК, что может вызвать самоуничтожение (апоптоз) определенных клеток, участвующих в развитии костей и тканей лица. Непонятно, почему эффекты снижения рРНК ограничиваются развитием лица. Мутации в TCOF1 и POLR1D вызывают аутосомно-доминантную форму Treacher Collins, а мутации в POLR1C вызывают аутосомно-рецессивную форму.
TCOF1 является первичным геном, связанным с TCS; мутация в этом гене обнаруживается у 90-95% людей с TCS. Однако у некоторых людей с типичными симптомами TCS мутации в TCOF1 не обнаружены. Исследование ДНК привело к идентификации мутаций, обнаруженных в TCOF1. Большинство мутаций представляют собой небольшие делеции или вставки, хотя также были идентифицированы сайт сплайсинга и миссенс-мутации.
Анализ мутаций выявил более 100 болезнетворных мутаций в TCOF1, которые в основном являются семейно-специфическими мутациями. На единственную рецидивирующую мутацию приходится около 17% случаев.
TCOF1 обнаруживается на 5-й хромосоме в области 5q32. Он кодирует относительно простой ядрышковый белок, называемый патока, который, как считается, участвует в сборке рибосом. Мутации в TCOF1 приводят к гаплонедостаточности белка патоки. Гаплонедостаточность возникает, когда диплоидный организм имеет только одну функциональную копию гена, потому что другая копия инактивирована мутацией. Одна нормальная копия гена не производит достаточного количества белка, что вызывает болезнь. Гаплонедостаточность белка патоки приводит к истощению предшественника клеток нервного гребня, что приводит к уменьшению количества клеток гребня, мигрирующих в первую и вторую глоточные дуги. Эти клетки играют важную роль в развитии черепно-лицевого внешнего вида, и потеря одной копии патоки влияет на способность клеток формировать кости и ткани лица.
Мутации POLR1C и POLR1D ответственны за меньшую часть случаев Treacher Collins. POLR1C находится на хромосоме 6 в положении 6q21.2, а POLR1D находится на хромосоме 13 в положении 13q12.2. Эти гены кодируют белковые субъединицы, общие для РНК-полимеразы I и III. Обе эти полимеразы важны для биогенеза рибосом.
TCS наследуется аутосомно-доминантным способом и пенетрантностью пораженный ген почти завершен. Однако некоторые недавние исследования описали некоторые редкие случаи, когда пенетрантность в TCS не была полной. Причинами могут быть переменная экспрессия, неполная пенетрантность или мозаицизм зародышевой линии. Только 40% мутаций передаются по наследству. Остальные 60% являются результатом мутации de novo, когда ребенок имеет новую мутацию в ответственном гене и не унаследовал ее ни от одного из родителей. В исходе болезни возникает межсемейная и внутрисемейная изменчивость. Это говорит о том, что при рождении пораженного ребенка важно обследовать родителей, чтобы определить, присутствует ли пораженный ген, поскольку у родителя может быть легкая форма заболевания, которая не была диагностирована. В этом случае риск рождения еще одного больного ребенка составляет 50%. Если у родителей нет пораженного гена, риск рецидива низок. В следующих поколениях тяжесть клинических симптомов увеличивается.
Мутации в основных генах, ответственных за TCS, могут быть обнаружены с помощью взятия проб ворсинок хориона или амниоцентеза. Эти методы не позволяют обнаружить редкие мутации. Ультрасонография может использоваться для выявления черепно-лицевых аномалий на более поздних сроках беременности, но может не выявить более легкие случаи.
TCS часто сначала подозревают при наличии характерных симптомов, наблюдаемых во время физического осмотра. Однако клинические проявления СТК могут напоминать другие заболевания, что затрудняет диагностику. Классификация OMENS была разработана как комплексный и поэтапный подход к дифференциации заболеваний. Этот акроним описывает пять различных проявлений дисморфологии, а именно орбитальную асимметрию, гипоплазию нижней челюсти, деформацию ушной раковины, развитие нервов и заболевание мягких тканей.
Орбитальная симметрия
Нижняя челюсть
ухо
лицевой нерв
мягкие ткани
Для подтверждения диагноза в TCS используется несколько методов. 99>
ортопантомограмма (OPG) - это панорамный рентгеновский снимок верхней и нижней челюсти. Он показывает двухмерное изображение от уха до уха. В частности, OPG способствует точному послеоперационному наблюдению и мониторингу роста костей при лечении с использованием моно- или двойного дистрактора. Таким образом, некоторые особенности TCS можно увидеть на OPG, но используются более совершенные методы, позволяющие охватить весь спектр аномалий TCS вместо того, чтобы показывать только аномалии челюсти.
Другой метод радиографической оценки - получение рентгеновского изображения всей головы. Боковая цефалометрическая рентгенограмма в TCS показывает гипоплазию лицевых костей, таких как скуловая кость, нижняя челюсть и сосцевидный отросток.
Наконец, затылочные рентгенограммы используются для обнаружения гипоплазии или нарушения целостности скуловой дуги.
КТ височной кости с использованием тонких срезов позволяет диагностировать степень стеноза и атрезии наружного слухового прохода, состояние полости среднего уха, отсутствие или диспластические и рудиментарные косточки, или аномалии внутреннего уха, такие как недостаточная улитка. Двух- и трехмерные КТ-реконструкции с VRT и покрытием костей и кожи полезны для более точной постановки и трехмерного планирования реконструктивной хирургии нижней челюсти и наружного уха.
Другие болезни имеют сходные характеристики с синдромом Тричера Коллинза. В дифференциальной диагностике следует учитывать акрофациальные дизостозы. Внешний вид лица напоминает синдром Тричера Коллинза, но у этих людей возникают дополнительные аномалии конечностей. Примерами этих заболеваний являются синдром Нагера и синдром Миллера.
При дифференциальной диагностике также следует учитывать окулоаурикуловертебральный спектр. Примером является гемифациальная микросомия, которая в первую очередь влияет на развитие уха, рта и нижней челюсти. Эта аномалия может возникать двусторонне. Еще одно заболевание, относящееся к этому спектру, - синдром Гольденхара, который включает в себя позвоночные аномалии, эпибульбарные дермоиды и деформации лица.
Лечение людей с TCS может включать вмешательство профессионалов из разных дисциплин. В первую очередь заботятся о дыхании и кормлении из-за гипоплазии нижней челюсти и обструкции гортани языком. Иногда им может потребоваться трахеостомия для поддержания проходимости дыхательных путей и гастростомия для обеспечения адекватного потребления калорий и защиты дыхательных путей. Корректирующая хирургия лица проводится в определенный возраст, в зависимости от состояния развития.
Обзор настоящих рекомендаций:
Потеря слуха при синдроме Тричера Коллинза вызвана деформацией структур в наружном и среднем ухе. Потеря слуха обычно двусторонняя с кондуктивной потерей около 50-70 дБ. Даже в случаях с нормальными ушными раковинами и открытыми наружными слуховыми проходами цепочка слуховых косточек часто деформируется.
Попытки хирургической реконструкции наружного слухового прохода и улучшения слуха у детей с СКС не дали положительных результатов.
Слуховая реабилитация с помощью слуховых аппаратов с костной фиксацией (BAHA) или обычных средств костной проводимости оказалась предпочтительнее хирургической реконструкции.
Заболевание может быть ассоциировано с рядом психологических симптомов, включая тревогу, депрессию, социальную фобию и беспокойство по поводу образа тела; люди также могут подвергаться дискриминации, издевательствам и обзываниям, особенно в молодом возрасте. Эти вопросы должны учитывать многопрофильная команда и родительская поддержка.
TCS встречается примерно у одного из 50 000 новорожденных в Европе. По оценкам, во всем мире он встречается от одного из 10 000 до 1 из 50 000 рождений.
Синдром назван в честь Эдварда Тричера Коллинза (1862–1932), англичанин хирург и офтальмолог, которые описали его основные черты в 1900 году. В 1949 году Адольф Франческетти и Дэвид Кляйн описали то же заболевание на основании своих собственных наблюдений как нижнечелюстно-лицевой дизостоз.. Термин нижнечелюстно-лицевой дизостоз используется для описания клинических проявлений.
Статья в New York Times, июль 1977 г. , перепечатанная в многочисленных газетах по всей стране в течение следующих недель. эта болезнь впервые привлекла внимание многих людей.
Беспорядок был показан в шоу Nip / Tuck, в эпизоде «Blu Mondae ». TLC «Рожденные без лица» с Джулианой Ветмор, которая родилась с самым тяжелым случаем в истории болезни этого синдрома, у которой отсутствуют 30-40% костей лица.
В 2010 году BBC Three документальный фильм Люби меня, люби мое лицо, освещал дело человека, Джоно Ланкастера, с этим состоянием. В 2011 году BBC Three вернулся к Джоно, чтобы осветить его и его партнершу Лору стремление создать семью в фильме «Так что, если мой ребенок родился как я?», Который впервые вышел в эфир в рамках сезона программ BBC Three о воспитании детей. Первый фильм был показан на BBC One незадолго до первой трансляции второго фильма на BBC Three. Третий фильм Ланкастера BBC Three, «В поисках семьи» на Facebook, в котором рассматривается усыновление, вышел в эфир в 2011 году.
В Wonder, детский роман, главный герой - ребенок с синдромом Тричера Коллинза. В ноябре 2017 года вышла экранизация 2017 года с Джулией Робертс, Оуэном Уилсоном и Джейкобом Тремблеем. Элисон Мидстокк, актриса и модель с этим заболеванием.
| Классификация | D |
|---|---|
| Внешние ресурсы |