
Войти
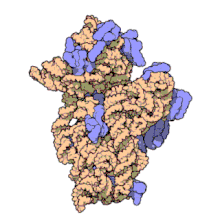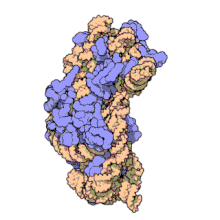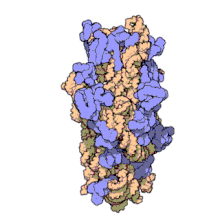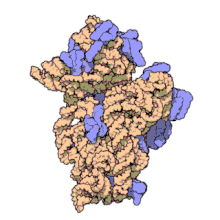 Молекулярная структура Субъединица 30S из Thermus thermophilus. Белки показаны синим, а одиночная цепь РНК - оранжевым.
Молекулярная структура Субъединица 30S из Thermus thermophilus. Белки показаны синим, а одиночная цепь РНК - оранжевым. 16S рибосомная РНК (или 16 S рРНК ) является компонентом 30S небольшой субъединицы прокариотическая рибосома, которая связывается с последовательностью Шайна-Далгарно. Кодирующие его гены называются геном 16S рРНК и используются для реконструкции филогении из-за медленных темпов эволюции этой области гена.. Карл Вёзе и Джордж Э. Фокс были двумя из тех, кто в 1977 году впервые применил 16S рРНК в филогенетике.
Множественные последовательности гена 16S рРНК могут существовать в пределах одной бактерии.
Он выполняет несколько функций:
Ген 16S рРНК используется для филогенетических исследований, поскольку он является высококонсервативным между различных видов бактерий и архей. Карл Вёзе (1977) впервые применил 16S рРНК. Предполагается, что ген 16S рРНК можно использовать в качестве надежных молекулярных часов, поскольку последовательности 16S рРНК от отдаленно родственных бактериальных линий, как показано, имеют сходные функциональные возможности. Некоторые термофильные археи (например, отряд Thermoproteales ) содержат ген 16S рРНК интроны, которые расположены в очень консервативные области и могут влиять на отжиг «универсальных» праймеров. Митохондриальная и хлоропластическая рРНК также амплифицируются.
Наиболее распространенная пара праймеров была разработана Weisburg et al. (1991) и в настоящее время обозначается как 27F и 1492R; однако для некоторых приложений могут потребоваться более короткие ампликоны, например, для секвенирования 454 с использованием химии титана пара праймеров 27F-534R, покрывающая от V1 до V3. Часто используется 8F, а не 27F. Два праймера почти идентичны, но 27F имеет M вместо C. AGAGTTTGATC M TGGCTCAG по сравнению с 8F.
| Название праймера | Последовательность (5′ – 3 ′) | Ссылка |
|---|---|---|
| 8F | AGA GTT TGA TCC TGG CTC AG | |
| 27F | AGA GTT TGA TC M TGG CTC AG | |
| U1492R | GGT TAC CTT GTT ACG ACT T | |
| 928F | TAA AAC TYA AAK GAA TTG ACG GG | |
| 336R | ACT GCT GCS YCC CGT AGG AGT CT | |
| 1100F | YAA CGA GCG CAA CCC | |
| 1100R | GGG TTG CGC TCG TTG | |
| 337F | GAC TCC TAC GGG AGG CWG CAG | |
| 907R | CCG TCA ATT CCT TTR AGT TT | |
| 785F | GGA TTA GAT ACC CTG GTA | |
| 805R | GAC TAC CAG GGT ATC TAA TC | |
| 533F | GTG CCA GCM GCC GCG GTA A | |
| 518R | GTA TTA CCG CGG CTG CTG G | |
| 1492R | CGG TTA CCT TGT TAC GAC TT |
Помимо высококонсервативных сайтов связывания праймеров, последовательности гена 16S рРНК содержат гипервариабельные области, которые могут обеспечивать видоспецифичные сигнатурные последовательности, полезные для идентификации бактерий. В результате секвенирование гена 16S рРНК стало распространенным в медицинской микробиологии как быстрая и дешевая альтернатива фенотипическим методам идентификации бактерий. Хотя первоначально оно использовалось для идентификации бактерий, впоследствии было обнаружено, что секвенирование 16S способно переклассифицировать бактерии в совершенно новые виды или даже роды. Он также использовался для описания новых видов, которые никогда не культивировались успешно. Поскольку секвенирование третьего поколения поступает во многие лаборатории, одновременная идентификация тысяч последовательностей 16S рРНК возможна в течение нескольких часов, что позволяет проводить метагеномные исследования, например, флоры кишечника.
Бактериальный ген 16S содержит девять гипервариабельных областей (V1 – V9) в диапазоне от 30 до 100 пар оснований длиной, которые участвуют во вторичной структуре малая рибосомная субъединица. Степень сохранения широко варьируется между гипервариабельными регионами, при этом более консервативные регионы соотносятся с таксономией более высокого уровня, а менее консервативные регионы - с более низкими уровнями, такими как род и виды. Хотя вся последовательность 16S позволяет сравнивать все гипервариабельные области, она имеет длину примерно 1500 пар оснований и может быть непомерно дорогостоящим для исследований, направленных на идентификацию или характеристику различных бактериальных сообществ. В этих исследованиях обычно используется платформа Illumina, которая производит считывания со скоростью в 50 и 12000 раз дешевле, чем 454 пиросеквенирование и секвенирование по Сэнгеру соответственно. Хотя секвенирование Illumina дешевле и обеспечивает более глубокий охват сообщества, оно дает считывания длиной только 75–250 пар оснований (до 300 пар оснований с Illumina MiSeq) и не имеет установленного протокола для надежной сборки полного гена в образцах сообщества. Однако полные гипервариабельные области могут быть собраны из одного прогона Illumina, что делает их идеальными мишенями для платформы.
Хотя гипервариабельные области 16S могут сильно различаться у разных бактерий, ген 16S в целом сохраняет большую однородность длины, чем его эукариотический аналог (18S рибосомная РНК ), который может упростить выравнивание. Кроме того, ген 16S содержит высококонсервативные последовательности между гипервариабельными областями, что позволяет создавать универсальные праймеры, которые могут надежно продуцировать одни и те же участки последовательности 16S в различных таксонах. Хотя ни одна гипервариабельная область не может точно классифицировать все бактерии от домена до видов, некоторые из них могут надежно предсказать конкретные таксономические уровни. Многие исследования сообщества выбирают полуконсервативные гипервариабельные области, такие как V4, по этой причине, поскольку они могут обеспечить разрешение на уровне филума так же точно, как и полный ген 16S. В то время как менее консервативные регионы изо всех сил пытаются классифицировать новые виды, когда таксономия более высокого порядка неизвестна, они часто используются для обнаружения присутствия конкретных патогенов. В одном исследовании Chakravorty et al. в 2007 году авторы охарактеризовали области V1 – V8 различных патогенов, чтобы определить, какие гипервариабельные области было бы наиболее полезно включать в специфические для болезни и широкие анализы. Среди других результатов они отметили, что область V3 лучше всего подходит для определения рода для всех протестированных патогенов, и что V6 является наиболее точным при дифференциации видов между всеми патогенами, наблюдаемыми под контролем CDC,, включая сибирскую язву..
Хотя анализ гипервариабельной области 16S является мощным инструментом для таксономических исследований бактерий, он изо всех сил пытается различить близкородственные виды. В семействах Enterobacteriaceae, Clostridiaceae и Peptostreptococcaceae виды могут иметь до 99% сходства последовательностей во всем гене 16S. В результате последовательности V4 могут отличаться всего на несколько нуклеотидов, в результате чего справочные базы данных не могут надежно классифицировать эти бактерии на более низких таксономических уровнях. Ограничивая анализ 16S для выбора гипервариабельных регионов, эти исследования могут не выявить различия в тесно связанных таксонах и сгруппировать их в единые таксономические единицы, что приведет к недооценке общего разнообразия выборки. Более того, бактериальные геномы могут содержать несколько генов 16S, причем области V1, V2 и V6 содержат наибольшее внутривидовое разнообразие. Хотя это и не самый точный метод классификации видов бактерий, анализ гипервариабельных областей остается одним из наиболее полезных инструментов, доступных для исследований бактериального сообщества.
При предположении, что эволюция управляется вертикальной передачей, гены 16S рРНК долгое время считались видоспецифичными и безошибочными в качестве генетических маркеров, определяющих филогенетические отношения между прокариотами. Однако растущее число наблюдений предполагает наличие горизонтального переноса этих генов. В дополнение к наблюдениям за естественным происхождением, переносимость этих генов подтверждается экспериментально с использованием специализированной генетической системы Escherichia coli. При использовании нулевого мутанта E. coli в качестве хозяина было показано, что рост мутантного штамма дополняется чужеродными генами 16S рРНК, которые филогенетически отличаются от E. coli на уровне филума. Такая функциональная совместимость также наблюдалась в Thermus thermophilus. Кроме того, у T. thermophilus наблюдали как полный, так и частичный перенос генов. Частичный перенос привел к спонтанному образованию явно случайной химеры между генами хозяина и чужеродными бактериальными генами. Таким образом, гены 16S рРНК могли развиваться посредством множества механизмов, включая вертикальное наследование и горизонтальный перенос генов ; частота последнего может быть намного выше, чем считалось ранее.
Ген 16S рРНК используется в качестве стандарта для классификации и идентификации микробов, поскольку он присутствует в большинстве микробов и показывает правильные изменения. Типовые штаммы последовательностей гена 16S рРНК для большинства бактерий и архей доступны в общедоступных базах данных, таких как NCBI. Однако качество последовательностей, найденных в этих базах данных, часто не проверяется. Поэтому широко используются вторичные базы данных, которые собирают только последовательности 16S рРНК. Наиболее часто используемые базы данных перечислены ниже:
База данных EzBioCloud, ранее известная как EzTaxon, состоит из полной иерархической таксономической системы, содержащей 62 988 видов бактерий и архей / phylotypes, который включает 15 290 действительных опубликованных названий по состоянию на сентябрь 2018 г. На основе филогенетических отношений, таких как максимальное правдоподобие и OrthoANI, все виды / подвиды представлены по крайней мере одной последовательностью гена 16S рРНК. База данных EzBioCloud систематически курируется и регулярно обновляется, что также включает новые виды-кандидаты. Кроме того, на веб-сайте представлены инструменты биоинформатики, такие как калькулятор ANI, ContEst16S и база данных рРНК 16S для QIIME и конвейера Mothur.
Проект базы данных рибосом (RDP) - это курируемая база данных, которая предлагает данные о рибосомах вместе с соответствующими программами и услугами. Предложения включают филогенетически упорядоченные выравнивания последовательностей рибосомной РНК (рРНК), производные филогенетические деревья, диаграммы вторичной структуры рРНК и различные пакеты программного обеспечения для обработки, анализа и отображения выравниваний и деревьев. Данные доступны по ftp и по электронной почте. Некоторые аналитические услуги также предоставляются сервером электронной почты.
SILVA предоставляет исчерпывающие, проверенные на качество и регулярно обновляемые наборы данных согласованных малых (16S / 18S, SSU) и больших подразделений (23S). / 28S, LSU) последовательности рибосомной РНК (рРНК) для всех трех доменов жизни, а также набор инструментов для поиска, создания праймеров и выравнивания (Bacteria, Archaea и Eukarya).
Greengenes - это комплексная справочная база данных и таксономия 16S с контролируемым качеством, основанная на филогении de novo, которая предоставляет стандартные операционные наборы таксономических единиц. Он больше не поддерживается активно и последний раз обновлялся в 2013 году.