
Войти
Секвенирование третьего поколения (также известное как секвенирование длительного чтения ) - это класс методов секвенирования ДНК в настоящее время активно разрабатываются.
Технологии секвенирования третьего поколения обладают способностью производить значительно более длинные чтения, чем секвенирование второго поколения. Такое преимущество имеет решающее значение как для науки о геноме, так и для изучения биологии в целом. Однако данные секвенирования третьего поколения имеют гораздо более высокий уровень ошибок, чем предыдущие технологии, что может усложнить последующую сборку генома и анализ полученных данных. Эти технологии активно развиваются, и ожидается, что они будут улучшены в отношении высокого уровня ошибок. Было обнаружено, что для приложений, которые более терпимы к частоте ошибок, таких как вызов структурных вариантов, секвенирование третьего поколения превосходит существующие методы.
Технологии секвенирования с подходом, отличным от платформ второго поколения, были впервые описаны как «третье поколение» в 2008 г. 2009.
В настоящее время в центре разработки технологий секвенирования третьего поколения находятся несколько компаний, а именно: Pacific Biosciences, Oxford Nanopore Technology, Quantapore (CA-USA) и Stratos (Вашингтон, США). Эти компании используют принципиально разные подходы к секвенированию отдельных молекул ДНК.
Компания PacBio разработала платформу секвенирования секвенирования одиночных молекул в реальном времени (SMRT), основанную на свойствах волноводов нулевого режима. Сигналы представляют собой флуоресцентное излучение света от каждого нуклеотида, включенного ДНК-полимеразой, связанной со дном лунки zL.
Технология Oxford Nanopore включает прохождение молекулы ДНК через наноразмерную структуру пор с последующим измерением изменений электрического поля, окружающего пору; в то время как Quantapore использует другой собственный подход к нанопорам. Stratos Genomics разделяет основания ДНК полимерными вставками, «Xpandomers», чтобы обойти проблему «сигнал-шум» при чтении нанопор оцДНК.
Также примечателен подход Helicos к флуоресценции одиночных молекул, но компания объявила о банкротстве осенью 2015 года.
По сравнению с технологиями секвенирования текущего поколения, секвенирование третьего поколения имеет очевидное преимущество, заключающееся в получении гораздо более длинных считываний. Ожидается, что такая большая длина чтения облегчит многочисленные вычислительные проблемы, связанные с сборкой генома, реконструкцией транскриптов и метагеномикой среди других важных областей современной биологии и медицины.
Хорошо известно, что геномы эукариот, включая приматов и людей, сложные и имеют большое количество длинных повторяющихся областей. Короткие чтения из секвенирования второго поколения должны прибегать к приближенным стратегиям, чтобы вывести последовательности на больших расстояниях для сборки и вызова генетических вариантов. Чтения конца пары были усилены секвенированием второго поколения для преодоления этих ограничений. Однако точные длины фрагментов на концах пары часто неизвестны и также должны быть приблизительно определены. Делая возможными длинные чтения, технологии секвенирования третьего поколения имеют явные преимущества.
Эпигенетические маркеры представляют собой стабильные и потенциально наследуемые модификации молекулы ДНК, которые не входят в ее последовательность. Примером является метилирование ДНК в сайтах CpG, которое, как было установлено, влияет на экспрессию генов. Другой пример - модификации гистонов. Текущее поколение технологий секвенирования основано на лабораторных методах, таких как ChIP-секвенирование для обнаружения эпигенетических маркеров. Эти методы включают маркировку нити ДНК, разрыв и фильтрацию фрагментов, содержащих маркеры, с последующим секвенированием. Секвенирование третьего поколения может позволить прямое обнаружение этих маркеров из-за их отличительного сигнала от других четырех нуклеотидных оснований.
 Портативный секвенсор генов MinION, Oxford Nanopore Technologies
Портативный секвенсор генов MinION, Oxford Nanopore Technologies Другие важные преимущества третьего технологии секвенирования поколения включают портативность и скорость секвенирования. Поскольку требуется минимальная предварительная обработка образцов по сравнению с секвенированием второго поколения, можно разработать оборудование меньшего размера. Компания Oxford Nanopore Technology недавно выпустила на рынок секвенсор MinION. Эта секвенсорная машина размером примерно с обычный USB-накопитель, и ее можно легко использовать, подключив к ноутбуку. Кроме того, поскольку процесс секвенирования не распараллеливается по участкам генома, данные можно собирать и анализировать в режиме реального времени. Эти преимущества секвенирования третьего поколения могут хорошо применяться в больничных условиях, где требуется быстрый сбор и анализ данных на месте.
Секвенирование третьего поколения в его нынешнем виде сталкивается с серьезными проблемами, в основном связанными с точной идентификацией нуклеотидных оснований; частота ошибок по-прежнему намного выше по сравнению с секвенированием второго поколения. Обычно это происходит из-за нестабильности задействованного молекулярного механизма. Например, в технологии одиночного молекулярного секвенирования и секвенирования в реальном времени PacBio молекула ДНК-полимеразы становится все более поврежденной по мере того, как происходит процесс секвенирования. Кроме того, поскольку процесс происходит быстро, сигналы отдельных баз могут быть размыты сигналами соседних баз. Это создает новую вычислительную задачу для расшифровки сигналов и, следовательно, определения последовательности. Такие методы, как скрытые марковские модели, например, использовались для этой цели с некоторым успехом.
В среднем разные люди в человеческой популяции имеют около 99,9% общих генов. Другими словами, примерно только одна из тысячи баз будет отличаться у любых двух человек. Высокая частота ошибок, связанная с секвенированием третьего поколения, неизбежно создает проблемы для характеристики индивидуальных различий, существующих между членами одного и того же вида.
Сборка генома представляет собой реконструкцию последовательностей ДНК целого генома. Обычно это делается с помощью двух принципиально разных подходов.
Когда доступен эталонный геном, как в случае человека, вновь секвенированные чтения можно просто выровнять с эталонным геномом, чтобы охарактеризовать его свойства. Такая сборка на основе ссылок является быстрой и простой, но ее недостатком является «сокрытие» новых последовательностей и вариантов с большим числом копий. Кроме того, для большинства организмов еще не существуют ссылочные геномы.
Сборка de novo - это альтернативный подход к сборке генома для эталонного выравнивания. Он относится к реконструкции последовательностей целого генома полностью из считанных исходных последовательностей. Этот метод будет выбран, когда эталонный геном отсутствует, когда вид данного организма неизвестно, как в метагеномике, или когда существуют представляющие интерес генетические варианты, которые не могут быть обнаружены путем сопоставления эталонного генома.
Учитывая короткие считывания, производимые текущим поколением технологий секвенирования, de novo сборка - серьезная вычислительная проблема. Обычно к ней прибегают с помощью итеративного процесса поиска и соединения считываний последовательностей с заметными перекрытиями. Различные вычислительные и статистические методы, такие как графы де Брюйна и согласованные графики пересечения макетов были использованы для решения этой проблемы. Тем не менее, из-за высокой повторяемости геномов эукариот, точная и полная реконструкция геномных последовательностей при сборке de novo остается сложной задачей. Считывание с конца пары было предложено как возможное решение, хотя точная длина фрагмента часто неизвестна и должна быть приближена.
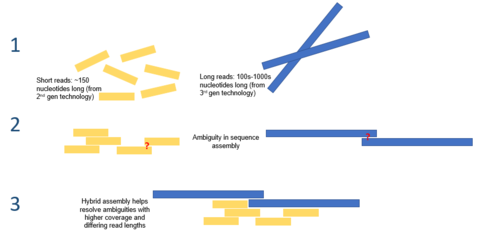 Гибридная сборка - использование считываний с платформ секвенирования 3-го поколения с короткими считываниями из 2-го поколения платформы - могут использоваться для устранения неоднозначностей, которые существуют в геномах, ранее собранных с использованием секвенирования второго поколения. Короткие чтения второго поколения также использовались для исправления ошибок, которые существуют в длинных считываниях третьего поколения.
Гибридная сборка - использование считываний с платформ секвенирования 3-го поколения с короткими считываниями из 2-го поколения платформы - могут использоваться для устранения неоднозначностей, которые существуют в геномах, ранее собранных с использованием секвенирования второго поколения. Короткие чтения второго поколения также использовались для исправления ошибок, которые существуют в длинных считываниях третьего поколения. Большая длина чтения, предлагаемая секвенированием третьего поколения, может облегчить многие из проблем, с которыми в настоящее время сталкивается de novo сборки генома. Например, если целая повторяющаяся область может быть однозначно упорядочена за одно считывание, никаких вычислений не потребуется. Вычислительные методы были предложены, чтобы облегчить проблему высокой частоты ошибок. Например, в одном исследовании было продемонстрировано, что сборка микробного генома de novo с использованием одного только секвенирования PacBio работает лучше, чем секвенирование второго поколения.
Секвенирование третьего поколения также может использоваться в сочетании с секвенированием второго поколения. Этот подход часто называют гибридным секвенированием. Например, длинные чтения из секвенирования третьего поколения могут использоваться для устранения неоднозначностей, которые существуют в геномах, ранее собранных с использованием секвенирования второго поколения. С другой стороны, короткие чтения второго поколения использовались для исправления ошибок, существующих в длинных чтениях третьего поколения. В целом было показано, что этот гибридный подход значительно улучшает сборки генома de novo.
метилирование ДНК (DNAm) - ковалентная модификация ДНК в CpG сайты, приводящие к присоединению метильных групп - это наиболее изученный компонент эпигенетического механизма. Модификации ДНК и результирующая экспрессия генов могут варьироваться в зависимости от типа клеток, временного развития, с генетическим происхождением, могут изменяться из-за воздействия окружающей среды и передаваться по наследству. После открытия ДНКm исследователи также обнаружили его связь с такими заболеваниями, как рак и аутизм. В контексте этиологии этого заболевания DNAm является важным направлением дальнейших исследований.
Текущие наиболее распространенные методы исследования состояния метилирования требуют анализа, который фрагментирует ДНК перед стандартным секвенированием второго поколения на платформе Illumina. В результате короткой длины чтения теряется информация о более длинных паттернах метилирования. Технологии секвенирования третьего поколения предлагают возможность секвенирования одной молекулы в реальном времени более длинных считываний и обнаружения модификации ДНК без вышеупомянутого анализа.
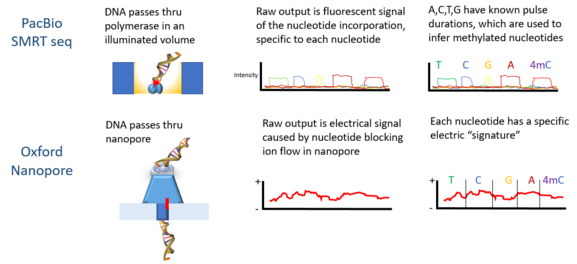 Технология PacBio SMRT и Oxford Nanopore могут использовать неизмененную ДНК для обнаружения метилирования.
Технология PacBio SMRT и Oxford Nanopore могут использовать неизмененную ДНК для обнаружения метилирования. Oxford Nanopore MinION был использован для обнаружения DNAm. Когда каждая нить ДНК проходит через пору, она производит электрические сигналы, которые, как было установлено, чувствительны к эпигенетическим изменениям в нуклеотидах, и скрытая марковская модель (HMM) использовалась для анализа данных MinION для обнаружения 5-метилцитозин (5mC) Модификация ДНК. Модель обучалась с использованием синтетически метилированного E. coli и полученные сигналы, измеренные с помощью нанопор. Затем обученную модель использовали для обнаружения 5mC в геномных считываниях MinION из линии клеток человека, которая уже имела эталонный метилом. Классификатор имеет точность 82% в случайно выбранных одноэлементных сайтах, которая увеличивается до 95% при применении более строгих пороговых значений.
Другие методы предназначены для различных типов модификаций ДНК с использованием платформы MinION. Stoiber et al. исследовали 4-метилцитозин (4mC) и 6-метиладенин (6mA) вместе с 5mC, а также создали программное обеспечение для прямой визуализации необработанных данных MinION удобным для человека способом. Здесь они обнаружили, что в E. coli, которая имеет известный метилом, окна событий длиной 5 пар оснований могут использоваться для разделения и статистического анализа необработанных электрических сигналов MinION. Простой U-тест Манна-Уитни может обнаруживать модифицированные части последовательности E. coli, а также дополнительно разбивать модификации на области 4mC, 6mA или 5mC.
Кажется вероятным, что в В будущем необработанные данные MinION будут использоваться для обнаружения множества различных эпигенетических меток в ДНК.
PacBio секвенирование также использовалось для обнаружения метилирования ДНК. В этой платформе ширина импульса - ширина импульса флуоресцентного света - соответствует определенной базе. В 2010 году было показано, что расстояние между импульсами в контрольных и метилированных образцах различно, и для каждого типа метилирования существует «сигнатурная» длительность импульса. В 2012 году с помощью платформы PacBio были охарактеризованы сайты связывания ДНК метилтрансфераз. Обнаружение N6-метилирования в C Elegans было показано в 2015 году. Метилирование ДНК по N-аденину с использованием платформы PacBio в эмбриональных стволовых клетках мыши было показано в 2016 году.
Другие формы модификаций ДНК - из-за тяжелых металлов, окисления или УФ-повреждения - также являются возможными направлениями исследований с использованием секвенирования третьего поколения Oxford Nanopore и PacBio.
Обработка необработанных данных - например, нормализация к среднему сигналу - была необходима для необработанных данных MinION, что ограничивало возможности технологии в реальном времени. Согласованность электрических сигналов все еще остается проблемой, что затрудняет точное определение нуклеотида. MinION имеет низкую пропускную способность; поскольку трудно получить множественные перекрывающиеся считывания, это дополнительно приводит к проблемам с точностью обнаружения последующих модификаций ДНК. Как скрытая марковская модель, так и статистические методы, используемые с необработанными данными MinION, требуют повторных наблюдений модификаций ДНК для обнаружения, а это означает, что отдельные модифицированные нуклеотиды должны постоянно присутствовать в нескольких копиях генома, например в нескольких клетках или плазмидах в образце.
Для платформы PacBio, в зависимости от того, какое метилирование вы ожидаете обнаружить, потребности в покрытии могут различаться. По состоянию на март 2017 года другие эпигенетические факторы, такие как модификации гистонов, не были обнаружены с помощью технологий третьего поколения. Более длинные паттерны метилирования часто теряются, потому что меньшие контиги все еще нуждаются в сборке.
Транскриптомика - это исследование транскриптома, обычно путем определения относительного количества молекул матричной РНК в исследуемой ткани. Согласно центральной догме молекулярной биологии, генетическая информация течет от двухцепочечных молекул ДНК к одноцепочечным молекулам мРНК, где они могут быть легко переведены в функциональные белковые молекулы. Изучая транскриптом, можно получить ценную информацию о регуляции экспрессии генов.
Хотя уровни экспрессии как уровень гена можно более или менее точно отобразить с помощью секвенирования второго поколения, информация об уровне транскрипта по-прежнему является важной проблемой. Как следствие, роль альтернативного сплайсинга в молекулярной биологии остается в значительной степени неуловимой. Технологии секвенирования третьего поколения открывают многообещающие перспективы в решении этой проблемы, позволяя секвенировать молекулы мРНК по всей их длине.
Альтернативный сплайсинг (AS) - это процесс, при котором один ген может давать начало множеству различных транскриптов мРНК и, следовательно, различным трансляциям белков. Некоторые данные свидетельствуют о том, что AS является повсеместным явлением и может играть ключевую роль в определении фенотипов организмов, особенно в сложных эукариотах; все эукариоты содержат гены, состоящие из интронов, которые могут подвергаться AS. В частности, было подсчитано, что AS встречается в 95% всех мультиэкзонных генов человека. AS имеет неоспоримый потенциал влиять на множество биологических процессов. Расширение знаний в этой области имеет решающее значение для изучения биологии в целом.
Текущее поколение технологий секвенирования производит только короткие чтения, что накладывает огромные ограничения на способность обнаруживать отдельные транскрипты; короткие чтения должны быть преобразованы в исходные расшифровки, которые могли бы дать повод для результирующих наблюдений чтения. Эта задача еще более усложняется из-за очень вариабельных уровней экспрессии в транскриптах и, как следствие, вариабельных покрытий чтения в последовательности гена. Кроме того, экзоны могут быть общими для отдельных транскриптов, что делает однозначные выводы практически невозможными. Существующие вычислительные методы делают выводы на основе накопления коротких чтений в различных местах последовательности, часто делая упрощающие предположения. Cufflinks использует экономный подход, стремясь объяснить все чтения с наименьшим возможным количеством расшифровок. С другой стороны, StringTie пытается одновременно оценить количество транскриптов во время сборки считываний. Эти методы, хотя и разумны, не всегда могут идентифицировать настоящие стенограммы.
В исследовании, опубликованном в 2008 году, было изучено 25 различных существующих протоколов реконструкции транскриптов. Его данные свидетельствуют о том, что существующие методы, как правило, неэффективны при сборке транскриптов, хотя способность обнаруживать отдельные экзоны относительно не нарушена. По оценкам, средняя чувствительность к обнаружению экзонов по 25 протоколам составляет 80% для генов Caenorhabditis elegans. Для сравнения, чувствительность идентификации транскриптов снижается до 65%. Для человека в исследовании сообщается, что чувствительность обнаружения экзонов в среднем составляет 69%, а чувствительность обнаружения транскриптов составляет в среднем всего 33%. Другими словами, для человека существующие методы способны идентифицировать менее половины всех существующих расшифровок.
Технологии секвенирования третьего поколения продемонстрировали многообещающие перспективы в решении проблемы обнаружения транскриптов, а также оценки количества мРНК на уровне транскриптов. Хотя уровень ошибок остается высоким, технологии секвенирования третьего поколения позволяют производить считывание гораздо большей длины. Pacific Bioscience представила платформу iso-seq, предлагая секвенировать молекулы мРНК по всей их длине. Ожидается, что Oxford Nanopore предложит аналогичные технологии. Проблема с более высоким коэффициентом ошибок может быть уменьшена за счет дополнительных высококачественных коротких чтений. Этот подход был ранее протестирован, и сообщалось, что он снижает частоту ошибок более чем в 3 раза.
Метагеномика - это анализ генетического материала, полученного непосредственно из образцов окружающей среды.
Основным преимуществом технологий секвенирования третьего поколения в метагеномике является их скорость секвенирования по сравнению с методами второго поколения. Скорость секвенирования важна, например, в клинических условиях (т. Е. идентификация патогена ), чтобы обеспечить эффективную диагностику и своевременные клинические действия.
MinION Oxford Nanopore использовался в 2015 году для метагеномного обнаружения патогенов в режиме реального времени в сложных клинических образцах с высоким уровнем фона. Первое считывание вируса Эбола (EBV) было секвенировано через 44 секунды после сбора данных. Было единое отображение считываний в геном; по крайней мере, одно чтение соответствует>88% генома. Относительно длинные считывания позволили секвенировать почти полный вирусный геном с высокой точностью (97–99% идентичности) непосредственно из первичного клинического образца.
Обычный филогенетический маркер микробного сообщества исследованиями разнообразия является ген 16S рибосомной РНК. Платформа SMRT MinION и PacBio использовалась для секвенирования этого гена. В этом контексте частота ошибок PacBio была сопоставима с частотой более коротких чтений с 454 и платформ секвенирования MiSeq компании Illumina.
Высокая частота ошибок MinION (~ 10-40 %) препятствует идентификации маркеров устойчивости к противомикробным препаратам, для которых необходимо разрешение единичных нуклеотидов. По той же причине не были идентифицированы эукариотические возбудители. Также вызывает беспокойство легкость переноса загрязнения при повторном использовании той же проточной кюветы (стандартные протоколы промывки не работают). Уникальные штрих-коды могут позволить большее мультиплексирование. Кроме того, выполнить точную видовую идентификацию для бактерий, грибов и паразитов очень сложно, поскольку они разделяют большую часть генома, а некоторые отличаются только <5%.
Стоимость базового секвенирования по-прежнему значительно выше, чем у MiSeq. Однако перспектива дополнения справочных баз данных полноразмерными последовательностями организмов ниже предела обнаружения с помощью подхода Sanger ; это могло бы очень помочь идентификации организмов в метагеномике.