
Войти
В химии, кинетический анализ хода реакций (РПКА ) представляет собой подмножество широкого диапазона кинетических методов, используемых для определения теории скорости химических средств и для помощи в использовании механизмов реакции. Хотя концепции, определяющие кинетический анализ реакций, не новы, этот процесс был формализован профессором Донной Блэкмонд (в настоящее время в Исследовательском институте Скриппса ) в конце 1990-х годов и с тех находит все более широкое распространение.. В отличие от более распространенного анализа псевдопервого порядка, в котором используется подавляющий избыток одного или нескольких реагентов по отношению к интересующим видам, РПКА исследует реакции в синтетически релевантных условиях (т. Е. При подходх и соотношениях реагентов, сходных с) те, которые используются в реакции, когда не исследуют закон скорости). Как правило, этот анализ включает систему, в которой проявляются различные реагенты, заметно изменяются в ходе реакций. механизм может действовать в зависимости от относительных и абсолютных концентраций задействованных веществ, этот подход дает результаты, которые гораздо более репрезентативны для поведения реакции в обычно используемых условиях, чем традиционные тактики. Кроме того, информация, полученная путем наблюдения за реакцией во времени, может дать представление о неожиданном поведении, таком как периоды индукции, дезактивация катализатора или изменения в механизме.
Кинетический анализ протекания реакции на способности точно контролировать конверсию реакции во времени. Эта цель может быть достигнута с помощью ряда методов, наиболее распространенные из способов ниже. Хотя эти методы иногда подразделяются на дифференциальные (мониторинг скорости во времени) или интегральные (мониторинг количества субстрата и продукта во времени), простые математические манипуляции (дифференциация или интеграция ) позволяет взаимное преобразование данных, полученных одним из двух. Независимо от применяемой техники, обычно подтверждать достоверность мониторинга с помощью дополнительного метода отслеживания.
ЯМР спектроскопия часто является методом выбора для мониторинга реакций, потребление субстрата и / или образование продукта можно наблюдать с течением времени по изменению интеграции пика относительно нереакционноспособного стандарта. Из данных о скорости реакции во времени может быть получена путем взятия производной от полиномиальной аппроксимации экспериментальной кривой. Ходовые реакции ЯМР можно классифицировать как интегральный метод, собранные первичные данные пропорциональны концентрации от времени. Он имеет недостаток, заключающийся в том, что требуется гомогенная система, поддающаяся реакции в трубке для ЯМР. Хотя наблюдение ЯМР может идентифицировать промежуточные продукты реакции, любого данного вида в ходе реакции не обязательно предполагает его в продуктивном процессе. Ход реакции ЯМР часто можно проводить при модели, позволяя регулировать скорость реакции до уровня, удобного для наблюдения. Примеры использования ЯМР реакции имеются в большом количестве, с яркими примерами, включая исследование аминирования Бухвальда-Хартвига (можно отметить, что значительная дискуссия окружала лучший подход к механистическому развитию аминирования Бухвальда-Хартвига, о чем свидетельствует число противоречивых) См. указанную статью и ссылки в ней.)
На месте инфракрасная спектроскопия может быть местечко для контроля за ходом реакции при условии, что реагент или продукт демонстрируют отличную оптическую плотность в ИК области качества. Скорость потребления реагента и / или продукта может быть отделена от изменения оптической плотности с течением времени (с применением закона Берса ). Даже когда спектры реагента и продукта в некоторой степени перекрываются, современное программное обеспечение для КИП способно точно деконволюционировать относительные вклады при условии резкого изменения обычно точно самого лучшего пика с течением времени. In situ IR можно классифицировать как интегральный метод, поскольку собираемые первичные данные пропорциональны концентрации от времени. Из этих данных можно получить исходный материал или концентрацию продукта с течением времени, просто взяв интеграл от полиномиальной аппроксимации экспериментальной кривой. С помощью методов доступности спектрометров с помощью мониторинга на месте, в последние годы наблюдается рост использования FT-IR. Примеры примечаний, включающих механистический анализ катализируемого амидотиомочевиной асимметричного синтеза Стрекера неприродных аминокислот и катализируемого основы Льюиса галолактонизация и циклоэтерификация.
Аналогично in situ ИК-экспериментам, описанным выше, in situ УФ-видимая спектроскопия усиленного может использоваться для мониторинга протекания реакции при условии, что реагент или продукт демонстрируют отличную оптическую плотность в УФ-области. Скорость потребления реагента и / или продукта может быть отделена от изменения оптической плотности с течением времени (с применением закона Бера ), что снова приводит к классификации в качестве интегрального метода. Из-за используемой спектральной области методы-видимого излучения чаще используются неорганические или металлоорганические системы, чем для исследования используются примеры использования самария реакции Барбье.
Калориметрия может установка для контроля за ходом реакции, мгновенный тепловой поток реакции, который связан с изменением реакции, контролируется. Реакционная калориметрия может быть классифицирована как дифференциальный метод, поскольку собранные первичные данные пропорциональны скорости в зависимости от времени. Из этих данных можно получить исходный материал или концентрацию продукта с течением времени, просто взяв интеграл от полиномиальной аппроксимации экспериментальной кривой. Хотя реакционная калориметрия используется реже, чем ряд других методов, она нашла применение в качестве эффективного инструмента для скрининга катализатора. Реакционная калориметрия также применяется как эффективный метод изучения механизмов механизмов, включая катализируемое пролинатом - катализируемое α- аминирование альдегидов и катализируемая палладием реакция аминирования Бухвальда-Хартвига.
Хотя Газовая хроматография, ВЭЖХ и масс-спектромет - все это отличные методы для различения смесей соединений (а иногда даже энантиомеров ), временное разрешение этих менее точно. чем у техник, описанных выше. Несмотря на это, эти методы все еще находят применение, например, при исследовании реакции Хека, где гетерогенный характер реакции не позволяет использовать методы, описанные выше. и SOMO-активация органокатализаторами. Несмотря на свои недостатки, эти методы могут служить отличными методами калибровки.
 a)Концентрация субстрата и продукта отслеживаются с течением времени такими методами, как in situ IR, UV-vis или ЯМР, или могут быть получены путем вычисления интеграла от (c). б) Представление дробного преобразования субстрата обеспечивает регулирование данных в (а). c) Скорость протекания реакции (образование продукта) отслеживает с течением времени с помощью таких методов, как калориметрия реакции, или ее можно получить, взяв первое производное от (а). d) Описание скорости распространения по распространению к потреблению исходного материала использует данные в более информативном распределении, чем наблюдаемое в (c). Обратите внимание, что ход реакции читается справа (максимальное значение субстрата) налево (без остатка субстрата) в (d).
a)Концентрация субстрата и продукта отслеживаются с течением времени такими методами, как in situ IR, UV-vis или ЯМР, или могут быть получены путем вычисления интеграла от (c). б) Представление дробного преобразования субстрата обеспечивает регулирование данных в (а). c) Скорость протекания реакции (образование продукта) отслеживает с течением времени с помощью таких методов, как калориметрия реакции, или ее можно получить, взяв первое производное от (а). d) Описание скорости распространения по распространению к потреблению исходного материала использует данные в более информативном распределении, чем наблюдаемое в (c). Обратите внимание, что ход реакции читается справа (максимальное значение субстрата) налево (без остатка субстрата) в (d). Данные о ходе реакции могут быть просто представлены в виде постоянной концентрации ([A] t) от времени (t) или дробного преобразования (F) от времени (t). Последнее требует незначительных алгебраических манипуляций для преобразования концентрации / преобразования в дробное преобразование (F) по:
где [A] 0 - количество, абсорбция или количество изначально присутствующего субстрата, а [A] t - количество, абсорбция, или это реагента в момент времени, т. Нормализация к дробному преобразованию может быть особенно полезной, поскольку она позволяет сравнивать несколько улучшений с разными абсолютными количествами или данными на одном графике.
Данные также обычно могут быть представлены в виде зависимости скорости (v) от времени (t). Опять же, требуются простые алгебраические манипуляции; например, калориметрические эксперименты дают:
, где q - мгновенная теплопередача, ΔH - известное изменение энтальпии реакции, а V представляет собой реакцию объем.
. Данные экспериментов по кинетике протекций реакции также часто предоставляют в виде графики зависимости скорости (v) от концентрации субстрата ([S]). Это требует получения и объединения графиков [S] в зависимости от t и v от t, описанных выше (обратите внимание, что один может быть получен из другого дифференцированного или интегрированного). Комбинация приводит к стандартному набору кривых, в котором Ход реакции считывается справа налево по оси x, а скорость реакции - снизу вверх по оси y. Хотя эти графики часто используют наглядную демонстрацию основных тенденций, статистические методы, как правило, лучше подходят для использования числовых констант скорости. (см. ниже)
В каталитической кинетике используются два основных приближения (в различных обстоятельствах) для описания поведения многих систем. Ситуации, которые могут быть связаны с помощью предварительного равновесия и стационарного состояния, часто можно использовать различные ситуации, связанные с состоянием покоя катализатора.
 a)В простейшем случае кинетики стационарного состояния одна подложка обратимо образует промежуточный комплекс (к используемому приближение стационарного состояния ) с катализатором, с последующим необратимым образованием продукта. b) В более сложном примере два субстрата связывают катализатор с последовательным образованием двух промежуточных соединений (и приближение стационарного состояния применяется к обоим) с последующим необратимым образованием продукта. Обратите внимание: поскольку I2 направляет непосредственно к свободному продукту и катализатору, это то же самое, что и в случае, когда I2 направляется непосредственно к свободному продукту и катализору. Для обоих (а) и (б) i) данный каталитический продукт с использованием константами скорости и весами, ii) отображает концентрацию продукта и реагента в ходе реакции., iii) как скорость реакции по мере того, как субстрат расходуется справа налево, и iv) показывает, что состояние покоя катализатора является полностью свободным катализатором, в то время как концентрация промежуточных продуктов остается небольшими и введенными, поскольку субстрат расходуется справа налево.
a)В простейшем случае кинетики стационарного состояния одна подложка обратимо образует промежуточный комплекс (к используемому приближение стационарного состояния ) с катализатором, с последующим необратимым образованием продукта. b) В более сложном примере два субстрата связывают катализатор с последовательным образованием двух промежуточных соединений (и приближение стационарного состояния применяется к обоим) с последующим необратимым образованием продукта. Обратите внимание: поскольку I2 направляет непосредственно к свободному продукту и катализатору, это то же самое, что и в случае, когда I2 направляется непосредственно к свободному продукту и катализору. Для обоих (а) и (б) i) данный каталитический продукт с использованием константами скорости и весами, ii) отображает концентрацию продукта и реагента в ходе реакции., iii) как скорость реакции по мере того, как субстрат расходуется справа налево, и iv) показывает, что состояние покоя катализатора является полностью свободным катализатором, в то время как концентрация промежуточных продуктов остается небольшими и введенными, поскольку субстрат расходуется справа налево. В стационарных условиях катализатор и субстрат подвергаются обратной ассоциации с последующим относительно быстрым расходом комплекс катализатор-субстрат (как прямая реакция на продукт, так и обратная реакция на несвязанный катализатор). приближение стационарного состояния утверждает, что на основе комплекса катализатор-субстрат не меняется время; общая конструкция системы остается низкой, так как он уносится почти сразу после образования. Закон скорости стационарного состояния содержит все константы скорости и вещества для использования от исходного материала к продукту, а знаменатель из суммы, описывающие описательные скорости прямых и обратных факторов, потребляющих промежуточное соединение в стационарном состоянии. В простейшем случае, когда один субстрат переходит в один продукт через одно промежуточное соединение:
В несколько более эффективная ситуация, когда два субстрата связываются последовательно с последующим высвобождением продукта:
Все более сложные системы могут быть описывается просто с помощью алгоритма, описанного в этой ссылке.
В случае стационарных условий, описанных выше, состояние покоя катализатора является несвязанной формой (промежуточное соединение, связанное с субстратом, по определению, присутствует только в при минимальной концентрации.)
 a)В простейшем случае предравновесной кинетики один субстрат быстро и обратимо образует промежуточный комплекс с катализатором с последующим необратимым образованием продукта. б) В более сложном примере два субстрата последовательно соединяют катализатор быстро и обратимо с последующим необратимым продуктом. Для обоих (а) и (б) i) данный каталитический продукт с использованием константами скорости и весами, ii) отображает концентрацию продукта и реагента в ходе реакции., iii) как скорость реакции по мере того, как субстрат расходуется справа налево, и iv) показывает, что состояние покоя катализатора представляет собой равновесное распределение свободного катализатора и промежуточных продуктов, где распределение все больше смещается в сторону свободного катализатора по мере того, как субстрат расходуется справа налево.
a)В простейшем случае предравновесной кинетики один субстрат быстро и обратимо образует промежуточный комплекс с катализатором с последующим необратимым образованием продукта. б) В более сложном примере два субстрата последовательно соединяют катализатор быстро и обратимо с последующим необратимым продуктом. Для обоих (а) и (б) i) данный каталитический продукт с использованием константами скорости и весами, ii) отображает концентрацию продукта и реагента в ходе реакции., iii) как скорость реакции по мере того, как субстрат расходуется справа налево, и iv) показывает, что состояние покоя катализатора представляет собой равновесное распределение свободного катализатора и промежуточных продуктов, где распределение все больше смещается в сторону свободного катализатора по мере того, как субстрат расходуется справа налево. В предварительных условиях равновесия катализатор и субстрат подвергаются быстрой реакции ассоциации перед медленной стадией, ведущей к образованию и высвобождению продукта. В этих условиях система может быть описана законом скорости "один плюс", где числитель состоит из всех констант скорости и веществ, необходимых для перехода от исходного материала к продукту, а знаменатель состоит из суммы, описывающих каждый из состояний, в которых существует катализатор ( а 1 соответствует свободному катализатору). В простейшем случае, когда один субстрат переходит в один продукт через одно промежуточное соединение:
В несколько более сложных ситуаций, когда два субстрата связываются последовательно с последующим высвобождением продукта:
В случае простых условий предварительного равновесия, описанных выше, состояние покоя катализатора или частично (в зависимости от величины константа равновесия) комплекс, связанный с субстратом.
Условия насыщения можно рассматривать как частный предварительный анализ равновесия. При исследуемой концентрации субстрата образования комплекса катализатор-субстрат происходит быстро и по существу необратимо. Состояние покоя катализатора полностью состоит из связанного комплекса, и [A] больше не присутствует в законе скорости; изменение [A] не повлияет на скорость реакции, поскольку катализатор уже полностью связан и реагирует так быстро, как позволяет k 2. Простейшим случаем кинетики насыщения является хорошо изученная модель кинетики ферментов Михаэлиса-Ментен.
 a)Простейший случай кинетики насыщения простирается от предравновесной ситуации, в которой быстрое комплексообразование субстрата с катализатором сопровождается медленным образованием продукта. b) Прямой участок графика концентрации субстрата с течением времени указывает на зависимость нулевого порядка от субстрата для большей части реакции, но кривая при низком [A] указывает на изменение на ( в данном случае) зависимость первого порядка от [A]. c) Насыщение катализатора очевидно при высоких концентрациях субстрата (где скорость не зависит от [A], но по мере расходования субстрата скорость реакции падает с зависимостью первого порядка от [A] ], чтобы пройти через источник. d) Состояние покоя катализатора также изменяется, где он существует почти полностью в виде связанного с субстратом комплекса, Кат-A, при высоком [A], но все чаще в виде свободного катализатора, Кат, поскольку [A] уменьшается в ходе реакции.
a)Простейший случай кинетики насыщения простирается от предравновесной ситуации, в которой быстрое комплексообразование субстрата с катализатором сопровождается медленным образованием продукта. b) Прямой участок графика концентрации субстрата с течением времени указывает на зависимость нулевого порядка от субстрата для большей части реакции, но кривая при низком [A] указывает на изменение на ( в данном случае) зависимость первого порядка от [A]. c) Насыщение катализатора очевидно при высоких концентрациях субстрата (где скорость не зависит от [A], но по мере расходования субстрата скорость реакции падает с зависимостью первого порядка от [A] ], чтобы пройти через источник. d) Состояние покоя катализатора также изменяется, где он существует почти полностью в виде связанного с субстратом комплекса, Кат-A, при высоком [A], но все чаще в виде свободного катализатора, Кат, поскольку [A] уменьшается в ходе реакции.  Скорость образования продукта при цианосилилировании кетона, A, показывает небольшую нелинейную зависимость от загрузки катализатора при высоких концентрациях катализатора. а) Это наблюдение, среди прочего, объясняется обратимым образованием неактивных каталитических комплексов. б) Подобное поведение в нескольких точках превращения согласуется с одним доминирующим механизмом, действующим на протяжении всего хода реакции.
Скорость образования продукта при цианосилилировании кетона, A, показывает небольшую нелинейную зависимость от загрузки катализатора при высоких концентрациях катализатора. а) Это наблюдение, среди прочего, объясняется обратимым образованием неактивных каталитических комплексов. б) Подобное поведение в нескольких точках превращения согласуется с одним доминирующим механизмом, действующим на протяжении всего хода реакции. Хотя реакция может демонстрировать один набор кинетических характеристик на ранней стадии превращения, это поведение может измениться из-за:
В случае кинетики насыщения, описанной выше, при условии, что [A] не присутствует в большом избытке по сравнению с [B], условия насыщения будут только применять в начале реакции. По мере того, как субстрат израсходован, концентрация уменьшается, и, в конце концов, [A] становится недостаточно, чтобы полностью подавить [Cat]. Это проявляется в постепенном изменении скорости от 0-го порядка к более высокому (например, 1-го, 2-го и т.д.) порядка в [A]. Это также можно описать как изменение состояния покоя катализатора от связанной формы к несвязанной форме в ходе реакции.
В дополнение к простому замедлению реакции изменение состояния покоя катализатора в ходе реакции может привести к появлению конкурирующих путей или процессов. Для доступа к продукту может присутствовать множество механизмов, и в этом случае порядок в катализаторе или субстрате может изменяться в зависимости от условий или точки реакции. Особенно полезный зонд для изменения механизма реакции включает изучение нормированной скорости реакции в зависимости от загрузки катализатора при множественных фиксированных точках превращения. Обратите внимание, что нормализованная скорость реакции:
корректируется с учетом расхода субстрата в ходе реакции, поэтому будут наблюдаться только изменения скорости из-за загрузки катализатора. Линейная зависимость от загрузки катализатора для даннойПреобразование зависимости на зависимость первого порядка от катализатора этой конверсии. Изменения линейности или нелинейности от одного набора превращения к другому указывает на изменения в зависимости от способа реакции. И наоборот, сохраняющихся в течение нескольких точек превращения (т.е. 30, 50 и 70%), указывает на изменение зависимости от катализатора на основе абсолютной изменения катализатора.
Взаимодействие катализатора с помощью компонентов реакционной смеси может привести к сложной кинетической зависимости. Хотя внецикловые взаимодействия катализатор-субстрат или катализатор-продукт обычно считаются «ядовитыми» для системы (конечно, в случае необратимого комплексообразования), существуют случаи, которые внецикловые частицы фактически защищают катализатор постоянной дезактивации. В любом случае важно понимать роль состояния покоя катализатора.
Переменным параметром, представляющим наибольший интерес для кинетического анализа хода реакции, избыток одного субстрата над другими в единицах молярности. Начальные частицы двух частиц в реакции могут быть оценены как:
и, предполагаемая, что - стехиометрия одной реакции, этот избыток одного субстрата над другим количественно сохраняется в течение всей реакции, так что:
Подобный набор может быть построен для условий со стехиометрией более высокого порядка, и в этом случае избыток предсказуемо изменяется в ходе реакции. Хотя е может быть положительным, отрицательным или нулевым, обычно в кинетическом анализе хода реакции используются положительные или отрицательные значения, меньшие по величине, чем один эквивалент субстрата. (Можно отметить, что кинетика псевдонулевого порядка использует избыточные значения, превышающие по величине один субстрата).
Определение фактора избытка (e) позволяет построить эксперименты с одинаковым избытком, в которых или более прогона кинетического эксперимента с разными начальными факторами, но одинаковый избыток позволяет искусственно войти в реакцию в любой момент. Эти эксперименты имеют решающее значение для RPKA каталитических возможностей, поскольку они позволяют исследовать ряд механистических возможностей, включая активацию катализатора (периоды индукции), дезактивацию катализатора и ингибирование продукта, которые более подробно обеспечивают ниже.
Перед дальнейшим изучением механизма определения кинетической зависимости интересующей реакции от катализатора. Частота оборота (TOF) катализатора может быть выражена как скорость реакции, нормированная на концентрацию катализатора:
Этот TOF определяется путем проведения любых двух или более экспериментов с одинаковым избытком. в котором лучшая конструкция катализатора рассматривается. Используют сертификат соответствия. Диагноз «совпадают». Здесь работают процессы более высокого порядка, которые требуют подробного анализа, описанные здесь. Также стоит отметить, что описанная здесь манипуляция нормализацией и наложением - только один из подходов к интерпретации необработанных данных. Столь же достоверные результаты могут быть получены путем подгонки наблюдаемого кинетического поведения к смоделированным законам скорости.
 a)Согласно предложенному механизму для катализируемого палладием аминирования арилгалогенидов, период индукции будет присутствовать, активный катализатор генерируется из неактивного предкатализатора (где L = BINAP ). b) Этот период индукции может наблюдаться в точках ранней конверсии, до того, как катализируемая реакция достигнет своей максимальной скорости. В экспериментах с одинаковым избытком это будет проявляться в неперекрываемых частях кривых, предназначенных для пересечения исходной реакции в промежуточных точках конверсии. Здесь одинаковый избыток (e = 0,60 M) [ArX] относительно [HNR 2 ] и [MOR] используется для каждой из кривых.
a)Согласно предложенному механизму для катализируемого палладием аминирования арилгалогенидов, период индукции будет присутствовать, активный катализатор генерируется из неактивного предкатализатора (где L = BINAP ). b) Этот период индукции может наблюдаться в точках ранней конверсии, до того, как катализируемая реакция достигнет своей максимальной скорости. В экспериментах с одинаковым избытком это будет проявляться в неперекрываемых частях кривых, предназначенных для пересечения исходной реакции в промежуточных точках конверсии. Здесь одинаковый избыток (e = 0,60 M) [ArX] относительно [HNR 2 ] и [MOR] используется для каждой из кривых. Как описано выше, такое же превышение экспериментов с двумя более экспериментами, поддерживающий избыток, (e) постоянным при изменении абсолютных концентраций субстратов (в этом случае катализатор также рассматривается как субстрат). Обратите внимание, что такая конструкция создает количество эквивалентов и, следовательно, молярный процент каждого реагента / катализатора должен различаться между реакциями. Эти эксперименты позволяют искусственно «войти» в реакцию в любой момент, благодаря исходной реакции одного эксперимента (реакция перехвата) выбираются таким образом, чтобы через привычный промежуточный момент времени в другом (исходная реакция).. Можно было бы описать, что ход реакции, подробно зависит от среды, содержащей субстрата, непосредственно друг на друга с этой перехвата точки. Однако это будет справедливо только в том случае, если скорость не изменится из-за изменений активности субстрата / катализатора (например, из-за активации катализатора, дезактивации катализатора или ингибирования продукта) перед этим перехватом.
Идеальное наложение нескольких экспериментов с одинаковым избытком, но разными начальными загрузками субстрата предполагает, что изменения в использовании активных субстрата / катализатора в ходе реакции не происходит. Отсутствие наложения графиков обычно указывает на активацию, дезактивацию катализатора или ингибирование продукта в условиях реакции. Эти случаи можно различить по положению кривых протекания реакции относительно друг друга. Перехваты использования, лежащих ниже (более медленных при той же концентрации субстрата) исходных факторов на графике зависимости скорости от концентрации субстрата, указывает на активацию катализатора в условиях реакции. Прерывание более высоких скоростей при той же концентрации субстрата) над исходными реакциями на графике зависимости скорости от концентрации субстрата, указывает на дезактивацию катализатора в условиях реакции; необходимы дальнейшие эксперименты, чтобы отличить ингибирование продукта от других форм гибели катализатора.
Одно из ключевых различий между реакцией перехвата и исходной точки, описанной выше, заключается в наличии некоторого количества продукта в исходной реакции в точке перехвата. Давно известно, что ингибирование продукта влияет на эффективность катализатора многих систем, и в случае экспериментов с одинаковым избытком оно предотвращает перекрывающую и родительскую реакцию. Хотя эксперименты с таким же избытком, как описано выше, не могут быть использованы для дезактивации катализатора какой-либо причиной, ингибирование продукта может быть исследовано с помощью дальнейших экспериментов, в которых некоторое начальное количество продукта добавлено к реакции перехвата (предназначено для имитации количества продукта, которое, как ожидается, должно присутствовать, исходной реакции при той же субстрата). Идеальное наложение графиков зависимости от скорости субстрата в условиях одинакового избытка и одинакового продукта указывает на то, что ингибирование продукта действительно происходит в условиях реакции. В то время как невозможность наложения графиков зависимости скорости от субстрата в условиях одинакового избытка и одного и того же препятствия ингибирования продукта, это, по крайней мере, указывает на то, что пути дезактивации катализатора также должны быть активными.
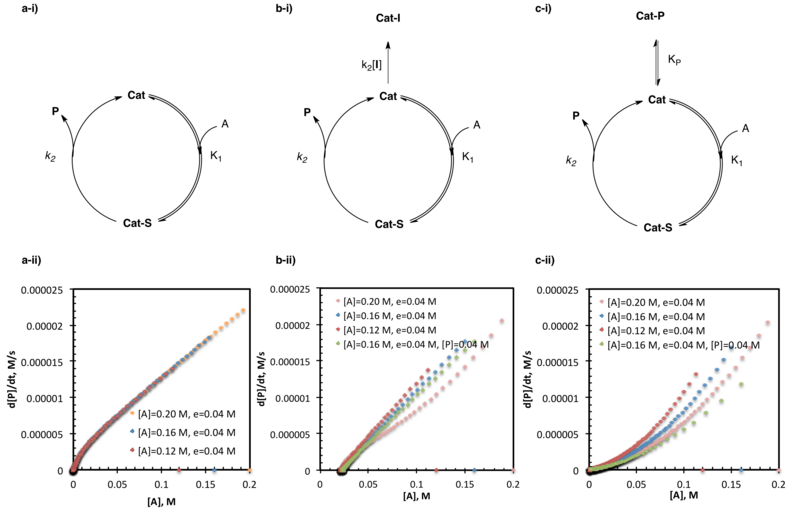 Кинетический анализ реакции может различить а) неингибированный катализатор б) необратимую гибель катализатора и в) ингибирование продукта посредством экспериментов с одинаковым избытком серии. Отсутствие показателей между уровнем и концентрацией субстрата для нескольких испытаний одной и той же реакции с одинаковым избытком, но разными начальными данными указывает на ингибирование или гибель катализатора. Их можно различить, где наложение экспериментов с одинаковым избытком с добавленным продуктом указывает на ингибирование продукта, где отсутствие наложения на альтернативную формуели гибели катализатора.
Кинетический анализ реакции может различить а) неингибированный катализатор б) необратимую гибель катализатора и в) ингибирование продукта посредством экспериментов с одинаковым избытком серии. Отсутствие показателей между уровнем и концентрацией субстрата для нескольких испытаний одной и той же реакции с одинаковым избытком, но разными начальными данными указывает на ингибирование или гибель катализатора. Их можно различить, где наложение экспериментов с одинаковым избытком с добавленным продуктом указывает на ингибирование продукта, где отсутствие наложения на альтернативную формуели гибели катализатора. Эксперименты с одинаковым избытком, исследующие дезактивацию катализатора и ингибирование продукта, наиболее широко используемые приложения кинетического анализа хода реакции. Среди примеров примеров в литературе некоторые исследования катализируемого аминоспиртом-алкилирования цинка альдегидов, катализируемого амидотиомочевиной асимметричного синтеза неприродных аминокислот и SOMO-активация органокатализаторов.
С большим количеством данных, доступных для мониторинга хода реакции во времени В сочетании с современными вычислительными методами стало довольно просто оцененно оценить скорости, сопоставив интегрированные законы скорости смоделированных вариантов закон с соответствием развития реакции времени. Благодаря использованию указанных выше методов, способам распространения ошибок, может быть увеличена неопределенность, чем построение графических уравнений скорости (см. Выше).
В то время как RPKA позволяет соблюдать скорость в течение всей реакции, проведения экспериментов только с одинаковым избытком не дает достаточной информации для определения соответствующих констант скорости. Чтобы построить независимые отношения для решения всех неизвестных констант скорости, необходимо исследовать систему с разным превышением.
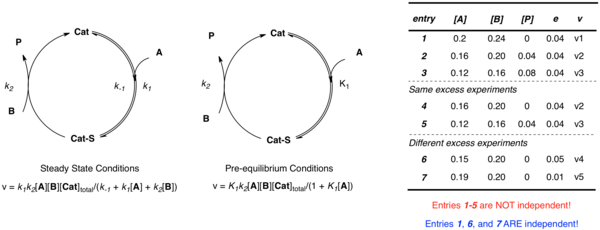 Рассмотрим простой пример, когда катализатор связывается с субстратом A, после чего следует реакция с продуктом B, P и свободным катализатором. Независимо от применяемого приближения, несколько независимых параметров (k 1, k −1 и k 2 в случае установившегося состояния; k 2 и K 1 в случае предварительного равновесия) требуются определения системы. Хотя можно представить себе несколько уравнений для описания неизвестных при различных экспериментах, когда данные получены из одинаковым избытком [A] и [B] не являются независимыми. Чтобы установить несколько уравнений, определяющих несколько независимых констант скорости с точки зрения экспериментальных скоростей и концентраций, необходимо несколько экспериментов с использованием различных значений е. Можно использовать нелинейный анализ методом наименьших квадратов для достижения наилучшего соответствия неизвестных констант этим уравнениям.
Рассмотрим простой пример, когда катализатор связывается с субстратом A, после чего следует реакция с продуктом B, P и свободным катализатором. Независимо от применяемого приближения, несколько независимых параметров (k 1, k −1 и k 2 в случае установившегося состояния; k 2 и K 1 в случае предварительного равновесия) требуются определения системы. Хотя можно представить себе несколько уравнений для описания неизвестных при различных экспериментах, когда данные получены из одинаковым избытком [A] и [B] не являются независимыми. Чтобы установить несколько уравнений, определяющих несколько независимых констант скорости с точки зрения экспериментальных скоростей и концентраций, необходимо несколько экспериментов с использованием различных значений е. Можно использовать нелинейный анализ методом наименьших квадратов для достижения наилучшего соответствия неизвестных констант этим уравнениям. Рассмотрим снова простой пример, обсужденный выше, где катализатор связывается с субстратом A, после чего следует реакция с B с образованием P и свободный катализатор. Независимо от применяемого приближения, несколько независимых параметров (k 2 и K 1 в случае предварительного равновесия; k 1, k −1 и k 2 в случае установившегося состояния) требуются для определения системы. Хотя можно представить себе построение нескольких уравнений для описания неизвестных при различных концентрациях, когда данные получены из эксперимента с одинаковым избытком [A] и [B] не являются независимыми:
Чтобы установить несколько независимых уравнений, определяющих несколько независимых констант скорости в терминах экспериментальных скоростей и концентраций, необходимо несколько экспериментов с разными значениями е. Затем можно использовать нелинейный анализ методом наименьших квадратов для получения значений наилучшего соответствия неизвестных констант скорости этим уравнениям.
 Одна возможная последовательность для установления кинетического порядка с помощью кинетического анализа хода реакции. Адаптировано с модификацией процесса, предложенного Донной Блэкмонд
Одна возможная последовательность для установления кинетического порядка с помощью кинетического анализа хода реакции. Адаптировано с модификацией процесса, предложенного Донной Блэкмонд Кинетики исторически полагались на линеаризацию данных скорости для экстраполяции констант скорости, что, возможно, лучше всего продемонстрировано широким использованием стандарта Лайнуивера – Берка линеаризация уравнения Михаэлиса – Ментен. Методы линеаризации имели особое значение до появления компьютерных методов, способных подбирать сложные кривые, и они остаются основным продуктом кинетики из-за их интуитивно простого представления. Важно отметить, что методы линеаризации НЕ должны использоваться для извлечения числовых констант скорости, поскольку они вносят большую степень ошибки по сравнению с альтернативными численными методами. Тем не менее, графические законы скорости поддерживают это интуитивное представление линеаризованных данных, так что визуальный осмотр графика может дать механистическое понимание реакции. В основе графического закона скорости лежит график зависимости скорости (v) от концентрации субстрата ([S]), обсуждаемый выше. Например, в простом цикле, обсуждаемом в отношении экспериментов с разным избытком, график зависимости v / [A] от [B] и его двойника v / [B] от [A] может дать интуитивное понимание порядка каждого реагентов. Если графики v / [A] по сравнению с [B] наложены для нескольких экспериментов с разным превышением, данные согласуются с зависимостью первого порядка от [A]. То же самое можно сказать и о сюжете v / [B] vs. [A]; наложение согласуется с зависимостью первого порядка от [B]. Возможны неперекрывающиеся результаты этих графических законов скорости, которые указывают на зависимость более высокого порядка от зондированных подложек. Блэкмонд предложила представить результаты экспериментов с помощью ряда графических уравнений (которые она представляет в адаптированной здесь блок-схеме), но важно отметить, что предложенный метод является лишь одним из многих методов отображения. кинетические отношения. Кроме того, представьте, что необходимо использовать для обработки данных и количественных констант скорости и порядков обработки..
Важно отметить, что даже несмотря на то, что кинетический анализ является важным инструментом для определения стехиометрии предельного переходного состояния по отношению к основному состоянию, он не может ответить на все механистические вопросы. Два механизма могут быть кинетически неразличимы, особенно в каталитических условиях. Для любой тщательной механистической оценки необходимо провести кинетический анализ как каталитического процесса, так и его отдельных этапов (когда это возможно) в сочетании с другими формами анализа, такими как оценка линейных зависимостей свободной энергии, исследования изотопного эффекта, вычислительный анализ или любое количество альтернативных подходов. Наконец, важно отметить, что никакая механистическая гипотеза никогда не может быть доказана; Альтернативная механистическая гипотеза может быть только опровергнута. Следовательно, важно проводить любое расследование, исходя из гипотез. Только