| МПЗ |
|---|
 |
| Доступные конструкции |
|---|
| PDB | Ортолог поиск: PDBe RCSB | | Список идентификационных кодов PDB |
|---|
3OAI | | |
| Идентификаторы |
|---|
| Псевдонимы | MPZ, CHM, CMT1, CMT1B, CMT2I, CMT2J, CMT4E, CMTDI3, CMTDID, DSS, HMSNIB, MPP, P0, нулевой миелиновый белок, CHN2 |
| Внешние идентификаторы | OMIM : 159440 MGI : 103177 HomoloGene : 445 GeneCard : MPZ |
|
| Расположение гена ( Мышь ) |
|---|
 | | Chr. | Хромосома 1 (мышь) |    | | Группа | 1 H3 | 1 79,05 см | Начинать | 171150711 п.н. | | Конец | 171 161 130 п.н. | |
|
|
| Ортологи |
|---|
| Разновидность | Человек | Мышь |
| Entrez | | |
| Ансамбль | | |
| UniProt | | |
| RefSeq (мРНК) | | |
| RefSeq (белок) | | |
| Расположение (UCSC) | Chr 1: 161.3 - 161.31 Мб | Chr 1: 171.15 - 171.16 Мб |
| PubMed поиск | | |
| Викиданные |
|
Нулевой миелиновый белок ( P0, MPZ) представляет собой одинарный мембранный гликопротеин, который у человека кодируется геном MPZ. Р0 является основным структурным компонентом миелиновой оболочки в периферической нервной системе (PNS). Нулевой миелиновый белок экспрессируется шванновскими клетками и составляет более 50% всех белков периферической нервной системы, что делает его наиболее распространенным белком, экспрессируемым в ПНС. Мутации в ноль белка миелина может вызвать дефицит миелина и связаны с невропатии, как болезнь Шарко-Мари-Тута и болезни Дежерин-Соттас.
СОДЕРЖАНИЕ
- 1 Структура
- 2 Функция
- 3 Связи с невропатией
- 4 ссылки
- 5 Дальнейшее чтение
- 6 Внешние ссылки
Состав
У человека ген, кодирующий нулевой миелиновый белок, расположен на хромосоме 1 рядом с локусом Даффи или антигеном / хемокиновым рецептором Даффи. Ген имеет длину около 7000 оснований и разделен на 6 экзонов. В общей сложности нулевой миелиновый белок имеет длину 219 аминокислот и много основных аминокислотных остатков.
Нулевой миелиновый белок состоит из внеклеточного N-концевого домена (аминокислоты 1–124), единственной трансмембранной области (125–150) и положительно заряженной внутриклеточной области меньшего размера (151–219). Его цитоплазматический домен сильно заряжен положительно, но предположительно не складывается в глобулярную структуру. Внеклеточный домен структурно подобен домену иммуноглобулина, и поэтому белок считается принадлежащим к суперсемейству иммуноглобулинов.
Помимо того, что он существует в качестве мономера, нулевой миелиновый белок, как известно, также образует димеры и тетрамеры с другими нулевыми молекулами миелинового белка у позвоночных.
Функция
Миелиновой оболочки представляет собой многослойную мембрану, уникальной для нервной системы, которая функционирует как изолятор, чтобы значительно увеличить скорость проведения импульса аксонов. Нулевой миелиновый белок, отсутствующий в центральной нервной системе, является основным компонентом миелиновой оболочки периферических нервов. Мутации, которые нарушают функцию нулевого белка миелина, могут привести к снижению экспрессии миелина и дегенерации миелиновой оболочки в периферической нервной системе. В настоящее время постулируется, что нулевая экспрессия миелинового белка вызывается сигналами от аксона. Однако более подробная информация о регуляции нулевого уровня миелинового белка неизвестна.
Предполагается, что нулевой миелиновый белок является структурным элементом в образовании и стабилизации миелина периферических нервов. Также предполагается, что нулевой миелиновый белок служит молекулой клеточной адгезии, удерживая вместе несколько слоев миелина. Когда миелинизирующая клетка несколько раз оборачивает свою мембрану вокруг аксона, генерируя несколько слоев миелина, нулевой миелиновый белок помогает удерживать эти слои компактными, выступая в качестве «клея», удерживающего слои миелина вместе. Он делает это, удерживая свою характерную спиралевидную структуру вместе за счет электростатических взаимодействий положительно заряженного внутриклеточного домена с кислыми липидами на цитоплазматической стороне противоположного бислоя. и за счет взаимодействия между гидрофобными глобулярными «головками» соседних внеклеточных доменов.
Функция нулевого белка миелина аналогична функции других белков с доменами иммуноглобина, таких как полииммуноглобин и белок Т4. Эти белки функционируют как связывающие и адгезионные молекулы и участвуют в гомотипических взаимодействиях или взаимодействиях, в которых участвуют два схожих белка. Нулевой миелиновый белок удерживает вместе миелиновую оболочку, участвуя в гомотипических взаимодействиях с другими нулевыми белками миелинового белка. Внеклеточный домен нулевого миелинового белка связывается с сфинголипидной мембраной миелина и удерживает вместе миелиновые слои, используя гомотипические взаимодействия с другими внеклеточными доменами нулевого миелинового белка и с внеклеточными остатками триптофана, взаимодействующими с мембраной.
Также было продемонстрировано, что нулевой миелиновый белок взаимодействует с другими белками, такими как периферический миелиновый белок 22. Однако на данный момент цель этих взаимодействий еще не определена.
Связь с невропатией
Известно, что мутации нулевого миелинового белка вызывают дегенерацию миелина и нейропатию. Считается, что мутации, которые снижают адгезионную функцию нулевого миелинового белка или его способность участвовать в гомеотипических взаимодействиях с другими нулевыми белками миелина, вызывают невропатию. Мутации нулевого белка миелина могут привести к проблемам с развитием миелина в раннем возрасте или к дегенерации миелина на аксоне в более позднем возрасте. Некоторые мутации могут вызывать невропатию в младенчестве, например болезнь Держерина-Соттаса, в то время как другие мутации могут вызывать невропатию в течение первых двух десятилетий жизни, как болезнь Шарко-Мари-Тута. Добавление заряженной аминокислоты или изменение остатка цистеина во внеклеточной мембране может привести к раннему началу невропатии. Усечение цитоплазматического домена или изменение третичной структуры нулевого миелинового белка также может привести к невропатии, поскольку было продемонстрировано, что цитоплазматический домен необходим для гомотипических взаимодействий.
использованная литература
дальнейшее чтение
- Патель П.И., Лупски-младший (апрель 1994 г.). «Болезнь Шарко-Мари-Тута: новая парадигма механизма наследственного заболевания». Тенденции в генетике. 10 (4): 128–33. DOI : 10.1016 / 0168-9525 (94) 90214-3. PMID 7518101.
- Роа BB, Лупски-младший (1995). «Молекулярная генетика невропатии Шарко-Мари-Тута». Успехи в генетике человека. 22. С. 117–52. DOI : 10.1007 / 978-1-4757-9062-7_3. ISBN 978-1-4757-9064-1. PMID 7762451. Отсутствует или пусто
|title=( справка ) - Нелис Э., Хайтес Н., Ван Брокховен С. (1999). «Мутации в генах периферического миелина и связанных генах при наследственных периферических невропатиях». Человеческая мутация. 13 (1): 11–28. DOI : 10.1002 / (SICI) 1098-1004 (1999) 13: 1 lt;11:: AID-HUMU2gt; 3.0.CO; 2-A. PMID 9888385.
- Ватанабе М., Ямамото Н., Окоши Н., Нагата Х., Коно Й., Хаяси А., Тамаока А., Сёдзи С. (сентябрь 2002 г.). «Кортикостероид-зависимая асимметричная нейропатия с нулевой мутацией гена миелинового белка». Неврология. 59 (5): 767–9. DOI : 10,1212 / wnl.59.5.767. PMID 12221176.
- Хаттори Н, Ямамото М., Йошихара Т., Коике Х., Накагава М., Йошикава Х, Охниши А., Хаясака К., Онодера О, Баба М., Ясуда Х, Сайто Т., Накашима К., Кира Дж, Кадзи Р., Ока Н., Собуэ Дж. (Январь 2003 г.). «Демиелинизирующие и аксональные особенности болезни Шарко-Мари-Тута с мутациями белков, связанных с миелином (PMP22, MPZ и Cx32): клинико-патологическое исследование 205 японских пациентов». Мозг. 126 (Pt 1): 134–51. DOI : 10,1093 / мозг / awg012. PMID 12477701.
- Shy ME (март 2006 г.). «Периферические невропатии, вызванные мутациями нулевого миелинового белка». Журнал неврологических наук. 242 (1–2): 55–66. DOI : 10.1016 / j.jns.2005.11.015. PMID 16414078. S2CID 32802793.
- Хаясака К., Нанао К., Тахара М., Сато В., Такада Дж., Миура М., Уэмура К. (октябрь 1991 г.). «Выделение и определение последовательности кДНК, кодирующей основной структурный белок периферического миелина человека». Сообщения о биохимических и биофизических исследованиях. 180 (2): 515–8. DOI : 10.1016 / S0006-291X (05) 81094-0. PMID 1719967.
- Оврие Р.А., Маклеод Дж. Г., Кончин Т. Э. (февраль 1987 г.). «Гипертрофические формы наследственной моторной и сенсорной нейропатии. Исследование гипертрофической болезни Шарко-Мари-Тута (HMSN типа I) и болезни Дежерина-Сотта (HMSN тип III) в детском возрасте». Мозг. 110 (Pt 1) (1): 121–48. DOI : 10,1093 / мозг / 110.1.121. PMID 3467805.
- Тачи Н., Исикава Ю., Минами Р. (1985). «Два случая врожденной нейропатии гипомиелинизации». Мозг и развитие. 6 (6): 560–5. DOI : 10.1016 / s0387-7604 (84) 80101-1. PMID 6099985. S2CID 4767216.
- Хаясака К., Химоро М., Ван И, Таката М., Миносима С., Симидзу Н., Миура М., Уэмура К., Такада Г. (сентябрь 1993 г.). «Структура и хромосомная локализация гена, кодирующего нулевой белок миелина человека (MPZ)». Геномика. 17 (3): 755–8. DOI : 10.1006 / geno.1993.1400. PMID 7503936.
- Су Й, Брукс Д. Г., Ли Л., Леперк Дж., Трофаттер Дж. А., Раветч Дж. В., Лебо Р. В. (ноябрь 1993 г.). «Ген нулевого белка миелина мутировал у пациентов типа 1B Шарко-Мари-Зуб». Труды Национальной академии наук Соединенных Штатов Америки. 90 (22): 10856–60. Bibcode : 1993PNAS... 9010856S. DOI : 10.1073 / pnas.90.22.10856. PMC 47877. PMID 7504284.
- Химоро М., Йошикава Х., Мацуи Т., Мицуи Ю., Такахаши М., Кайдо М., Нисимура Т., Савайши И., Такада Г., Хаясака К. (сентябрь 1993 г.). «Новая мутация гена миелина P0 в родословной нейропатии Шарко-Мари-Тута 1». Международная биохимия и молекулярная биология. 31 (1): 169–73. PMID 7505151.
- Хаясака К., Химоро М., Савайши И., Нанао К., Такахаши Т., Такада Г., Николсон Г.А., Оврие Р.А., Тачи Н. (ноябрь 1993 г.). «Мутация de novo гена миелина P0 при болезни Дежерина-Соттаса (наследственная моторная и сенсорная нейропатия III типа)». Генетика природы. 5 (3): 266–8. DOI : 10.1038 / ng1193-266. PMID 7506095. S2CID 2512684.
- Pham-Dinh D, Fourbil Y, Blanquet F, Mattéi MG, Roeckel N, Latour P, Chazot G, Vandenberghe A, Dautigny A (декабрь 1993 г.). «Главный нулевой ген периферического миелинового белка: структура и локализация в кластере генов рецепторов Fc гамма на хромосоме 1q21.3-q23 человека». Молекулярная генетика человека. 2 (12): 2051–4. DOI : 10.1093 / HMG / 2.12.2051. PMID 7509228.
- Томас Ф. П., Лебо Р. В., Росоклия Г., Динг XS, Лавлейс Р. Э., Латов Н., Хейс А. П. (1994). «Томакулезная невропатия в хромосоме 1 синдром Шарко-Мари-Тута». Acta Neuropathologica. 87 (1): 91–7. DOI : 10.1007 / BF00386259. PMID 7511317. S2CID 19827120.
- Нелис Э., Тиммерман В., Де Йонге П., Ванденберге А., Фам-Динь Д., Даутиньи А., Мартин Дж. Дж., Ван Брокховен С. (декабрь 1994 г.). «Быстрый скрининг генов миелина у пациентов с CMT1 с помощью анализа SSCP: выявление новых мутаций и полиморфизмов в гене P0». Генетика человека. 94 (6): 653–7. DOI : 10.1007 / bf00206959. PMID 7527371. S2CID 5750189.
- Hilmi S, Fournier M, Valeins H, Gandar JC, Bonnet J (февраль 1995 г.). «Гликопротеин миелина P0: идентификация сайта, фосфорилированного in vitro и in vivo эндогенными протеинкиназами». Журнал нейрохимии. 64 (2): 902–7. DOI : 10.1046 / j.1471-4159.1995.64020902.x. PMID 7530295. S2CID 32511382.
- Раутенштраус Б., Нелис Э., Грел Х., Пфайфер Р.А., Ван Брокховен С. (сентябрь 1994 г.). «Идентификация инсерционной мутации de novo в P0 у пациента с фенотипом синдрома Дежерина-Соттаса (DSS)». Молекулярная генетика человека. 3 (9): 1701–2. DOI : 10.1093 / HMG / 3.9.1701. PMID 7530550.
- Латур П., Бланке Ф., Нелис Э., Боннебуш С., Чапон Ф., Дирасон П., Олланьон Э., Даутиньи А., Фам-Динь Д., Шазо Г. (1995). «Мутации в гене нулевого белка миелина, связанные с болезнью Шарко-Мари-Тута типа 1B». Человеческая мутация. 6 (1): 50–4. DOI : 10.1002 / humu.1380060110. PMID 7550231. S2CID 20852048.
внешние ссылки
Эта статья включает текст из общественного достояния
Pfam и
InterPro :
IPR019566

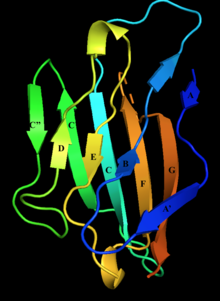 Структура внеклеточного домена нулевого миелинового белка с мечеными бета-цепями. Нити D, E, B и A составляют один бета-лист, нити A ', G, F, C, C', C '' составляют другой бета-лист.
Структура внеклеточного домена нулевого миелинового белка с мечеными бета-цепями. Нити D, E, B и A составляют один бета-лист, нити A ', G, F, C, C', C '' составляют другой бета-лист.

