
Войти
| Поликистоз почек | |
|---|---|
| Другие названия | Почки - поликистоз |
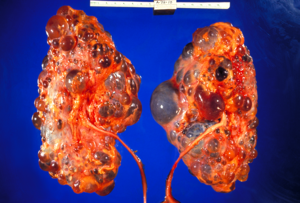 | |
| Поликистоз почек с тяжелым поражением удален в время трансплантации | |
| Специальность | Нефрология |
| Симптомы | Боль в животе |
| Типы | ADPKD и ARPKD |
| Метод диагностики | МРТ, КТ, УЗИ |
| Лечение | Антигипертензивные средства, управление образом жизни |
Поликистоз почек (PKD или PCKD, также известный как синдром поликистозных почек ) представляет собой генетическое заболевание, при котором почечные канальцы становятся структурно аномальными, что приводит к развитию и росту множественных кист внутри почки. Эти кисты могут начать развиваться в утробе матери, в младенчестве, в детстве или во взрослом возрасте. Кисты - это нефункционирующие канальцы, заполненные закачанной в них жидкостью, размер которых варьируется от микроскопических до огромных, разрушающих соседние нормальные канальцы и, в конечном итоге, делая их нефункциональными.
PKD вызывается аномальными генами, которые продуцируют специфический аномальный белок; этот белок отрицательно влияет на развитие канальцев. PKD - это общий термин для двух типов, каждый из которых имеет свою патологию и генетическую причину: аутосомно-доминантная поликистозная болезнь почек (ADPKD) и аутосомно-рецессивная поликистозная болезнь почек (ARPKD). Аномальный ген существует во всех клетках тела; в результате кисты могут образовываться в печени, семенных пузырьках и поджелудочной железе. Этот генетический дефект также может вызывать аневризмы корня аорты и аневризмы в Уиллисовском круге церебральных артерий, которые в случае их разрыва могут вызвать субарахноидальную оболочку. кровоизлияние.
Диагноз можно заподозрить на основании одного, некоторых или всех следующих факторов: впервые возникшая боль в боку или красная моча; положительный семейный анамнез; пальпация увеличенных почек при физикальном осмотре; случайная находка на УЗИ брюшной полости ; или случайное обнаружение аномальной функции почек при рутинной лабораторной работе (АМК, креатинин сыворотки или рСКФ ). Окончательный диагноз ставится при обследовании КТ брюшной полости.
Осложнения включают гипертензию из-за активации ренин-ангиотензин-альдостероновой системы (РААС), частые кистозные инфекции, мочевые кровотечения и снижение функции почек. Гипертонию лечат ингибиторами ангиотензинпревращающего фермента (ИАПФ ) или блокаторами рецепторов ангиотензина (БРА). Инфекции лечат антибиотиками. Снижение функции почек лечится с помощью заместительной почечной терапии (ЗПТ): диализа и / или трансплантации. С момента подозрения или постановки окончательного диагноза лечение осуществляет сертифицированный нефролог.
Признаки и симптомы включают высокое кровяное давление, головные боли, боли в животе, кровь в моче и чрезмерное мочеиспускание. Другие симптомы включают боль в спине и образование кист (почек и других органов).
PKD вызывается аномальными генами, которые продуцируют специфический аномальный белок, который оказывает неблагоприятное воздействие на канальцы. развитие. PKD - это общий термин для двух типов, каждый из которых имеет свою патологию и генетическую причину: аутосомно-доминантная поликистозная болезнь почек (ADPKD) и аутосомно-рецессивная поликистозная болезнь почек (ARPKD).
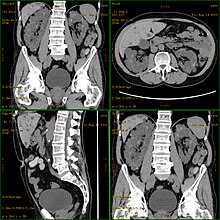 КТ, показывающий аутосомно-доминантную поликистозную болезнь почек
КТ, показывающий аутосомно-доминантную поликистозную болезнь почек  Карикатура аутосомно-доминантной поликистозной болезни почек с нормальной вставкой почек справа от диаграммы
Карикатура аутосомно-доминантной поликистозной болезни почек с нормальной вставкой почек справа от диаграммы  Мультфильм аутосомно-рецессивной поликистозной болезни почек с нормальной вставкой почек справа от диаграммы
Мультфильм аутосомно-рецессивной поликистозной болезни почек с нормальной вставкой почек справа от диаграммы Аутосомно-доминантная поликистозная болезнь почек (АДПБП) является наиболее распространенной из всех наследственных кистозных болезней почек с частотой 1: 500 живорожденных. Исследования показывают, что у 10% пациентов с терминальной стадией заболевания почек (ESKD), проходящих лечение диализом в Европе и США, изначально диагностировали и лечили ADPKD.
Генетически мутации в любом из трех генов PKD1, PKD2 и имеют сходные фенотипические проявления.
Аутосомно-рецессивный поликистоз почек (ARPKD) (OMIM # 263200) является менее распространенным из двух типов PKD, с частотой 1: 20 000 живорождений и обычно выявляется в первые несколько недель после рождения. К сожалению, почки часто недоразвиты, что приводит к 30% смертности новорожденных с ARPKD. PKHD1 вовлечен.
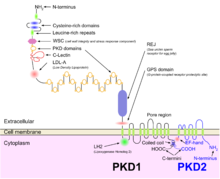 PKD1 и PKD2
PKD1 и PKD2 Формирование кисты как аутосомно-доминантной, так и аутосомно-рецессивной поликистозной болезни почек связано с аномальными ресничками -опосредованными сигнализация. Белки полицистин-1 и полицистин-2, по-видимому, вовлечены как в аутосомно-доминантную, так и в рецессивную поликистозную болезнь почек из-за дефектов обоих белков. Оба белка взаимодействуют с белками кальциевых каналов и вызывают снижение уровня покоящегося (внутриклеточного) кальция и накопления кальция в эндоплазматическом ретикулуме.
Заболевание характеризуется феноменом «второго удара», при котором мутировавший доминантный аллель унаследовано от родителя, причем образование кисты происходит только после того, как нормальный ген дикого типа выдерживает последующий второй генетический «удар», что приводит к образованию кисты почечных канальцев и прогрессированию заболевания.
PKD возникает в результате дефектов в первичная ресничка, неподвижная волосовидная клеточная органелла, присутствующая на поверхности большинства клеток тела, закрепленная в теле клетки за счет базального тела. В почках первичные реснички присутствуют на большинстве клеток нефрона, выступая с апикальной поверхности почечного эпителия в просвет канальцев. Считалось, что реснички изгибаются в потоке мочи, что приводит к изменениям в передаче сигналов, однако с тех пор было показано, что это экспериментальная ошибка (изгиб ресничек было артефактом компенсации фокальной плоскости, а также фактическим влиянием на мочеиспускание серьезным воздействием на мочеиспускание. гипертония и остановка сердца) и что изгиб ресничек не способствует изменению потока кальция. Хотя неизвестно, как дефекты первичной реснички приводят к развитию кисты, считается, что это может быть связано с нарушением одного из многих сигнальных путей, регулируемых первичной ресничкой, включая внутриклеточный кальций, Wnt / β-катенин, циклический аденозин. монофосфат (цАМФ) или планарная клеточная полярность (PCP). Нарушается функция первичной реснички, что приводит к нарушению ряда внутриклеточных сигнальных каскадов, которые вызывают дифференцировку кистозного эпителия, усиление деления клеток, усиление апоптоза и потерю способности к резорбции.
Поликистоз почек можно определить с помощью компьютерной томографии брюшной полости, а также МРТ и УЗИ той же области. Медицинский осмотр / тест может выявить увеличенную печень, шумы в сердце и повышенное кровяное давление
Большинство случаев прогрессируют до двустороннего заболевания в зрелом возрасте..
 Chr 11 FISH-сопоставленные BAC от CGAP
Chr 11 FISH-сопоставленные BAC от CGAP Нет одобренного FDA лечения. Однако недавние исследования показывают, что умеренные или умеренные диетические ограничения замедляют прогрессирование аутосомно-доминантной поликистозной болезни почек (ADPKD) у мышей.
Если и когда заболевание прогрессирует достаточно в данном случае, нефролог или другой практикующий врач и пациент должны решить, какая форма заместительной почечной терапии будет использоваться для лечения терминальной стадии заболевания почек (почечная недостаточность, обычно стадия 4 или 5 хронического заболевания почек).
Это будет либо какая-то форма диализа, который можно проводить двумя разными способами с разной частотой и продолжительностью (будь то выполняются дома или в клинике, зависит от используемого метода, устойчивости и подготовки пациента) и, в конечном итоге, если они соответствуют критериям в силу характера и тяжести их состояния и если можно найти подходящее соответствие, одностороннее или двустороннее трансплантация почки.
Исследование аутосомно-доминантного поликистоза почек в Кокрановском обзоре Легкость отметила тот факт, что важно всегда, избегая устойчивости к антибиотикам, контролировать инфекции кист в почках и, если поражены, печени, когда это необходимо в течение определенного времени для борьбы инфекция при использовании «бактериостатических и бактерицидных препаратов».
Люди с ADPKD могут вести нормальную жизнь; и наоборот, ARPKD может вызвать дисфункцию почек и может привести к почечной недостаточности к 40–60 годам. ADPKD1 и ADPKD2 сильно различаются тем, что ADPKD2 намного мягче.
В настоящее время не существует методов лечения, доказавших свою эффективность для предотвращения прогрессирования ADPKD.
PKD - это одно из самых распространенных наследственных заболеваний в США, от которого страдают более 600 000 человек. Это причина почти 10% всех терминальных стадий почечной недостаточности. Это одинаково влияет на мужчин, женщин и всех рас. PKD встречается у некоторых животных, а также у людей.
| Классификация | D |
|---|---|
| Внешние ресурсы |