
Войти
| Пропионовая ацидемия | |
|---|---|
| Другие названия | Гиперглицинемия с кетоацидозом и лейкопенией |
 | |
| Пропионовая кислота | |
| Специальность | Эндокринология |
| Симптомы | Низкий мышечный тонус, летаргия, рвота |
| Метод диагностики | Генетическое тестирование; высокий уровень пропионовой кислоты в моче |
| Лечение | Низкобелковая диета |
| Прогноз | Развитие может быть нормальным, или у пациентов может быть нарушение обучаемости на протяжении всей жизни |
Пропионовая ацидемия, также известная как пропионовая ацидурия или дефицит пропионил-КоА-карбоксилазы (дефицит PCC ), является редким аутосомно-рецессивным нарушение обмена веществ, классифицируемое как разветвленная цепь органическая ацидемия.
Расстройство проявляется в раннем неонатальном периоде с плохим питанием, рвотой, летаргией и отсутствие мышечного тонуса. Без лечения смерть может наступить быстро из-за вторичной гипераммонемии, инфекции, кардиомиопатии или повреждения головного мозга.
Пропионовая ацидемия почти сразу же характерна для новорожденных. Симптомы включают плохое питание, рвоту, обезвоживание, ацидоз, низкий мышечный тонус (гипотония ), судороги, и летаргия. Последствия пропионовой ацидемии быстро становятся опасными для жизни.
Долгосрочные осложнения могут включать хроническое заболевание почек, кардиомиопатию и удлинение интервала QTc.
 Метилмалоновая ацидемия вызывается недостатком витамина B 12 -зависимый фермент метилмалонил-КоА мутаза.
Метилмалоновая ацидемия вызывается недостатком витамина B 12 -зависимый фермент метилмалонил-КоА мутаза.  Пропионовая ацидемия имеет аутосомно-рецессивный характер наследования.
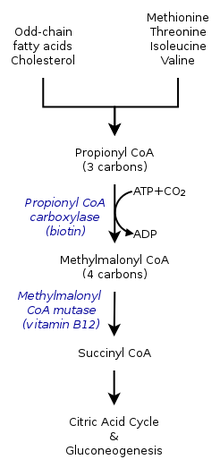
Пропионовая ацидемия имеет аутосомно-рецессивный характер наследования.У здоровых людей фермент пропионил-КоА-карбоксилаза превращает пропионил-КоА в метилмалонил -CoA. Это один из многих этапов процесса преобразования определенных аминокислот и жиров в энергию. Люди с пропионовой ацидемией не могут выполнить это преобразование, потому что фермент пропионил-КоА-карбоксилаза нефункционален. Незаменимые аминокислоты валин, метионин, изолейцин и треонин не могут быть преобразованы, и это приводит к накоплению пропионил-КоА. Вместо того, чтобы превращаться в метилмалонил-КоА, пропионил-КоА затем превращается в пропионовую кислоту, которая накапливается в кровотоке. Это, в свою очередь, вызывает накопление опасных кислот и токсинов, которые могут вызвать повреждение органов.
Во многих случаях пропионовая ацидемия может поражать мозг, сердце, почки, печень, вызывать судороги и задерживать нормальное развитие. например, ходьба или разговор. Пациенту может потребоваться госпитализация, чтобы предотвратить расщепление белков в организме. Необходимо тщательно контролировать диетические потребности.
Мутации в обеих копиях генов PCCA или PCCB вызывают пропионовую ацидемию. Эти гены содержат инструкции по формированию альфа- и бета-субъединиц PCC, фермент, называемый пропионил-CoA-карбоксилаза.
PCC, необходим для нормального расщепления незаменимых аминокислот валина, изолейцина, треонин и метионин, а также некоторые жирные кислоты с нечетной цепью. Мутации в генах PCCA или PCCB нарушают функцию фермента, предотвращая метаболизм этих кислот. В результате пропионил-КоА, пропионовая кислота, кетоны, аммиак и другие токсичные соединения накапливаются в крови., вызывающая признаки и симптомы пропионовой ацидемии.
Повышенное содержание метаболитов пропионовой кислоты (например, 3-гидроксипропионат, метилцитрат, тиглилглицин, пропионилглицин) в крови и моче во время с нормальным уровнем биотинидазы.
Пациентам с пропионовой ацидемией следует как можно раньше начать диету с низким содержанием белка. В дополнение к смеси белков, не содержащей метионина, треонина, валина и изолейцина, пациенту также следует получать лечение L-карнитином и давать антибиотики 10 дней в месяц для удаления пропиогенной флоры кишечника.. Пациенту должны быть составлены протоколы диеты: «дневная диета» с низким содержанием белка, «полуэкстренная диета», содержащая половину потребности в белке, и «экстренная диета» без содержания белка. Эти пациенты подвержены риску тяжелой гипераммонемии во время инфекций, которые могут привести к коматозным состояниям.
Трансплантат печени приобретает все большую роль в ведении таких пациентов, при этом небольшие серии показывают улучшение качества жизни.
Пропионовая ацидемия наследуется по аутосомной рецессивной модели и встречается примерно у 1 из 35000 живорождений в United Состояния. Это заболевание чаще встречается в Саудовской Аравии, с частотой примерно 1 из 3000. Это состояние также часто встречается у амишей, меннонитов и других групп населения с более высокой частотой кровного родства.
В 1957 году мужчина ребенок родился с плохим умственным развитием, повторяющимися приступами ацидоза и высоким уровнем кетонов и глицина в крови. После диетического тестирования доктор Бартон Чайлдс обнаружил, что его симптомы ухудшались при введении аминокислот лейцина, изолейцина, валина, метионина и треонина. В 1961 году медицинская бригада больницы Джонса Хопкинса в Балтиморе, Мэриленд опубликовала случай, назвав расстройство кетотической гиперглицинемией . В 1969 году, используя данные сестры первоначального пациента, ученые установили, что пропионовая ацидемия является рецессивным заболеванием и что пропионовая ацидемия и метилмалоновая ацидемия вызваны недостаточностью одного и того же ферментативного пути.
| Классификация | D |
|---|---|
| Внешние ресурсы |