
Войти
| Наследственная спастическая параплегия | |
|---|---|
| Специальность | Неврология |
Наследственная спастическая параплегия (HSP ) - это группа наследственных заболеваний, основной особенностью которых является прогрессирующее нарушение походки. Заболевание проявляется прогрессирующей скованностью (спастичность ) и сокращением в нижних конечностях. HSP также известен как наследственный спастический парапарез, семейная спастическая параплегия, болезнь французского поселения, болезнь Стрампелла или болезнь Стрампелла-Лоррена. Симптомы являются результатом дисфункции длинных аксонов в спинном мозге. Пораженные клетки представляют собой первичные двигательные нейроны ; следовательно, заболевание является заболеванием верхнего двигательного нейрона. HSP не является формой церебрального паралича, хотя физически может проявляться и вести себя так же, как спастическая диплегия. Происхождение HSP отличается от церебрального паралича. Несмотря на это, некоторые из тех же антиспастических препаратов, которые используются при спастическом церебральном параличе, иногда используются для лечения симптомов HSP.
HSP вызывается дефектами транспорта белков, структурных белков, белков, поддерживающих клетку, липидов и других веществ через клетку. Поражаются длинные нервные волокна (аксоны), потому что большие расстояния делают нервные клетки особенно чувствительными к дефектам в упомянутых механизмах.
Заболевание было впервые описано в 1880 году немецким неврологом Адольфом Штрюмпеллом. Более подробно он был описан в 1888 году французским врачом Морисом Лорреном. Благодаря их вкладу в описание болезни, во франкоязычных странах ее до сих пор называют болезнью Штрюмпеля-Лоррена. Термин «наследственная спастическая параплегия» был придуман Анитой Хардинг в 1983 году.
Симптомы зависят от типа унаследованного HSP. Основным признаком заболевания является прогрессирующая спастичность в нижних конечностях вследствие дисфункции пирамидного тракта. Это также приводит к оживленным рефлексам, подошвенным разгибателям, мышечной слабости и различным нарушениям мочевого пузыря. Кроме того, к основным симптомам HSP также относятся аномальная походка и трудности при ходьбе, снижение чувствительности к вибрации в лодыжках и парестезия. Люди с HSP могут испытывать сильную усталость, связанную с центральной нервной системой и нервно-мышечными расстройствами, которые могут приводить к инвалидности. Начальными симптомами обычно являются трудности с равновесием, ушибление пальца ноги или спотыкание. Симптомы HSP могут появиться в любом возрасте, от младенчества до старше 60 лет. Если симптомы появляются в подростковом возрасте или позже, то спастическое нарушение походки обычно прогрессирует в течение многих лет. Со временем могут потребоваться трости, ходунки и инвалидные коляски, хотя некоторым людям не требуются вспомогательные приспособления. Инвалидность описывается как более быстрое прогрессирование у взрослых форм.
Более конкретно, у пациентов с аутосомно-доминантной чистой формой HSP обнаруживаются нормальные движения лица и глазного яблока. Хотя подергивание челюсти может быть резким у пожилых людей, нарушения речи или затруднения глотания отсутствуют. Мышечный тонус и сила верхних конечностей в норме. В нижних конечностях мышечный тонус повышен в подколенных сухожилиях, четырехглавой мышце и лодыжках. Слабость наиболее заметна в подвздошно-поясничной мышце, передней большеберцовой мышце и, в меньшей степени, мышцах задней поверхности бедра. При сложной форме расстройства присутствуют дополнительные симптомы. К ним относятся: периферическая невропатия, амиотрофия, атаксия, умственная отсталость, ихтиоз, эпилепсия, оптическая невропатия, деменция, глухота или проблемы с речью, глотанием или дыханием.
Анита Хардинг классифицировала HSP как чистая и сложная форма. Чистый HSP проявляется спастичностью в нижних конечностях, связанной с нейрогенным нарушением мочевого пузыря, а также отсутствием чувствительности к вибрации (паллгипестезия ). С другой стороны, HSP классифицируется как комплексный, когда спастичность нижних конечностей сочетается с любыми дополнительными неврологическими симптомами.
Эта классификация является субъективной, и у пациентов со сложными HSP иногда диагностируется мозжечковая атаксия со спастичностью, умственная отсталость (со спастичностью) или лейкодистрофия. Некоторые из перечисленных ниже генов были описаны ранее при других заболеваниях, кроме HSP. Таким образом, некоторые ключевые гены перекрываются с другими группами болезней.
В прошлом HSP классифицировали как раннее начало, начинающееся в раннем детстве или более позднее начало в зрелом возрасте. Возраст начала имеет две максимальные точки - в возрасте 2 лет и около 40 лет. Новые данные показывают, что более раннее начало приводит к более длительной продолжительности заболевания без потери возможности передвигаться или необходимости использования инвалидной коляски. Ранее было также описано, что формы с более поздним началом развиваются быстрее.
HSP - это группа генетических нарушений. Он подчиняется общим правилам и может наследоваться по типу аутосомно-доминантным, аутосомно-рецессивным или X-сцепленным рецессивным. Участвующий способ наследования имеет прямое влияние на шансы унаследовать заболевание. Было описано более 70 генотипов, и более 50 генетических локусов были связаны с этим заболеванием. Было идентифицировано десять генов с аутосомно-доминантным наследованием. На один из них, SPG4, приходится ~ 50% всех генетически решенных случаев, или примерно 25% всех случаев HSP. Известно, что двенадцать генов наследуются по аутосомно-рецессивному типу. В совокупности последняя группа составляет ~ 1/3 случаев.
Большинство измененных генов имеют известную функцию, но для некоторых функция еще не идентифицирована. Все они перечислены в списке генов ниже, включая способ наследования. Некоторыми примерами являются спастин (SPG4) и параплегин (SPG7), оба являются АТФазами AAA.
Гены обозначены SPG (спастическая походка ген). Расположение генов имеет формат: хромосома - плечо (короткое или p: длинное или q) - номер полосы. Эти обозначения предназначены только для генов человека. Расположение может (и, вероятно, будет) отличаться у других организмов. Несмотря на количество известных генов, участвующих в этом состоянии, ~ 40% случаев еще не идентифицированы. В таблице ниже SPG? используется для обозначения гена, который был связан с HSP, но еще не получил официального обозначения гена HSP.
| Генотип | OMIM | Символ гена | Генный локус | Наследование | Возраст начала | Другие названия и характеристики |
|---|---|---|---|---|---|---|
| SPG1 | 303350 | L1CAM | Xq28 | Х-сцепленный рецессивный | Ранний | синдром MASA |
| SPG2 | 312920 | PLP1 | Xq22.2 | Х-сцепленный рецессивный | Переменная | Болезнь Пелицея-Мерцбахера |
| SPG3A | 182600 | ATL1 | 14q22.1 | Аутосомно-доминантный | Ранний | Болезнь Стрампелла (эта Wiki) |
| SPG4 | 182601 | SPAST | 2p22.3 | Аутосомно-доминантный | Переменная | |
| SPG5A | 270800 | CYP7B1 | 8q12.3 | Аутосомно-рецессивный | Переменная | |
| SPG6 | 600363 | NIPA1 | 15q11.2 | Аутосомно-доминантный | Переменная | |
| SPG7 | 607259 | SPG7 | 16q24.3 | Аутосомно-доминантный | Переменная | |
| SPG8 | 603563 | KIAA0196 | 8q24.13 | Аутосомно-доминантный | Взрослый | |
| SPG9A | 601162 | ALDH18A1 | 10q24.1 | Аутосомно-доминантный | Подростковый | Катаракта с двигательной нейронопатией, низким ростом и аномалиями скелета |
| SPG9B | 616586 | ALDH18A1 | 10q24.1 | Аутосомно рецессивный | Ранний | |
| SPG10 | 604187 | KIF5A | 12q13.3 | Аутосомно-доминантный | Ранний | |
| SPG11 | 604360 | SPG11 | 15q21.1 | Аутосомно-рецессивный | Переменная | |
| SPG12 | 604805 | RTN2 | 19q13.32 | Аутосомно-доминантный | Ранний | |
| SPG13 | 605280 | HSP60 | 2q33.1 | Аутосомно-доминантный | Переменная | |
| SPG14 | 605229 | ? | 3q27 – q28 | Аутосомно-рецессивный | Взрослый | |
| SPG15 | 270700 | ZFYVE26 | 14q24.1 | Аутосомно-рецессивный | Ранний | |
| SPG16 | 300266 | ? | Xq11.2 | Х-сцепленный рецессивный | Ранний | |
| SPG17 | 270685 | BSCL2 | 11q12. 3 | Аутосомно-доминантный | Подростковый | |
| SPG18 | 611225 | ERLIN2 | 8p11.23 | Аутосомно-рецессивный | Раннее | |
| SPG19 | 607152 | ? | 9q | Аутосомно-доминантное | Взрослое начало | |
| SPG20 | 275900 | SPG20 | 13q13.3 | Аутосомно-рецессивный | Раннее начало | Синдром Тройера |
| SPG21 | 248900 | ACP33 | 15q22.31 | Аутосомно-рецессивный | Раннее начало | синдром MAST |
| SPG22 | 300523 | SLC16A2 | Xq13.2 | Х-сцепленный рецессивный | Раннее начало | Синдром Аллана-Херндона-Дадли |
| SPG23 | 270750 | RIPK5 | 1q32.1 | Аутосомно-рецессивный | Раннее начало | Синдром Лисона |
| SPG24 | 607584 | ? | 13q14 | Аутосомно-рецессивный | Раннее начало | |
| SPG25 | 608220 | ? | 6q23 – q24.1 | Аутосомно-рецессивный | Взрослый | |
| SPG26 | 609195 | B4GALNT1 | 12q13.3 | Аутосомно-рецессивный | Раннее начало | |
| SPG27 | 609041 | ? | 10q22.1 – q24.1 | Аутосомно-рецессивный | Переменная | |
| SPG28 | 609340 | DDHD1 | 14q22.1 | Аутосомный рек эссивное | Раннее начало | |
| SPG29 | 609727 | ? | 1p31.1 – p21.1 | Аутосомно-доминантное | Подростковое | |
| SPG30 | 610357 | KIF1A | 2q37.3 | Аутосомно-рецессивный | Подростковый | |
| SPG31 | 610250 | REEP1 | 2p11.2 | Аутосомно доминантный | Раннее начало | |
| SPG32 | 611252 | ? | 14q12 – q21 | Аутосомно-рецессивный | Детство | |
| SPG33 | 610244 | ZFYVE27 | 10q24.2 | Аутосомно-доминантный | Взрослый | |
| SPG34 | 300750 | ? | Xq24 – q25 | Х-сцепленный рецессивный | Подростковый / Взрослый | |
| SPG35 | 612319 | FA2H | 16q23.1 | Аутосомно-рецессивный | Детство | |
| SPG36 | 613096 | ? | 12q23– q24 | Аутосомно-доминантный | Подростковый / Взрослый | |
| SPG37 | 611945 | ? | 8p21.1 – q13.3 | Аутосомно-доминантный | Переменная | |
| SPG38 | 612335 | ? | 4p16 – p15 | Аутосомно-доминантный | Подростковый / взрослый | |
| SPG39 | 612020 | PNPLA6 | 19p13.2 | Аутосомно-рецессивный | Детство | |
| SPG41 | 613364 | ? | 11p14.1 – p11.2 | Аутосомно-доминантный | Подростковый возраст | |
| SPG42 | 612539 | SLC33A1 | 3q25.31 | Аутосомно-доминантный | Переменная | |
| SPG43 | 615043 | C19orf12 | 19q12 | Аутосомно-рецессивный | Детство | |
| SPG44 | 613206 | GJC2 | 1q42.13 | Аутосомно-рецессивный | Детство / подросток | |
| SPG45 | 613162 | NT5C2 | 10q24.32 – q24.33 | Аутосомно-рецессивный | Младенческий | |
| SPG46 | 614409 | GBA2 | 9p13.3 | Аутосомно-рецессивный | Переменная | |
| SPG47 | 614066 | AP4B1 | 1p13.2 | Аутосомно-рецессивный | Детство | |
| SPG48 | 613647 | AP5Z1 | 7p22.1 | Аутосомно-рецессивный | 6-я декада | |
| SPG49 | 615041 | TECPR2 | 14q32.31 | Аутосомно-рецессивный | Младенческий | |
| SPG50 | 612936 | AP4M1 | 7q22.1 | Аутосомно-рецессивный | Младенчество | |
| SPG51 | 613744 | AP4E1 | 15q21.2 | Аутосомно-рецессивный | Младенческий | |
| SPG52 | 614067 | AP4S1 | 14q12 | Аутосомно-рецессивный | Младенчество | |
| SPG53 | 614898 | VPS37A | 8p22 | Аутосомно-рецессивный | Детство | |
| SPG54 | 615033 | DDHD2 | 8p11.23 | Аутосомно-рецессивный | Детство | |
| SPG55 | 615035 | C12orf65 | 12q24.31 | Аутосомно-рецессивный | Детство | |
| SPG56 | 615030 | CYP2U1 | 4q25 | Аутосомно-рецессивный | Детство | |
| SPG57 | 615658 | TFG | 3q12.2 | Аутосомно-рецессивный | Ранний | |
| SPG58 | 611302 | KIF1C | 17p13.2 | Аутосомно-рецессивный | В течение первых двух десятилетий | |
| SPG59 | 603158 | USP8 | 15q21.2 | ? Аутосомно-рецессивный | Детство | |
| SPG60 | 612167 | 3p22.2 | ? Аутосомно-рецессивный | Младенческий возраст | ||
| SPG61 | 615685 | ARL6IP1 | 16p12.3 | Аутосомные рецессы ve | Младенчество | |
| SPG62 | 615681 | ERLIN1 | 10q24.31 | Аутосомно-рецессивный | Детство | |
| SPG63 | 615686 | AMPD2 | 1p13.3 | Аутосомно-рецессивный | Младенческий | |
| SPG64 | 615683 | ENTPD1 | 10q24.1 | Аутосомно-рецессивный | Детство | |
| SPG66 | 610009 | 5q32 | ? Аутосомно-доминантное | Младенчество | ||
| SPG67 | 615802 | PGAP1 | 2q33.1 | Аутосомно-рецессивный | Младенческий | |
| SPG68 | 609541 | KLC2 | 11q13.1 | Аутосомно-рецессивный | Детство | Синдром SPOAN |
| SPG69 | 609275 | RAB3GAP2 | 1q41 | Аутосомно-рецессивный | Младенчество | , Микросиндром Варбурга |
| SPG70 | 156560 | MARS | 12q13 | ? Аутосомно-доминантный | Младенческий | |
| SPG71 | 615635 | ZFR | 5p13.3 | ? Аутосомно-рецессивный | Детство | |
| SPG72 | 615625 | REEP2 | 5q31 | Аутосомно-рецессивный;. аутосомно-доминантный | Инфанц y | |
| SPG73 | 616282 | CPT1C | 19q13.33 | Аутосомно-доминантный | Взрослый | |
| SPG74 | 616451 | IBA57 | 1q42.13 | Аутосомно-рецессивный | Детство | |
| SPG75 | 616680 | MAG | 19q13.12 | Аутосомно-рецессивный | Детство | |
| SPG76 | 616907 | CAPN1 | 11q13 | Аутосомно-рецессивный | Взрослый | |
| SPG77 | 617046 | FARS2 | 6p25 | Аутосомно-рецессивный | Детство | |
| SPG78 | 617225 | ATP13A2 | 1p36 | Аутосомно-рецессивный | Взрослый | Куфор –Синдром Ракеба |
| SPG79 | 615491 | UCHL1 | 4p13 | Аутосомно-рецессивный | Детство | |
| HSNSP | 256840 | CCT5 | 5p15.2 | Аутосомно-рецессивный | Детство | со спастической параплегией |
| SPG? | SERAC1 | 6q25.3 | Ювенильный | |||
| SPG? | 605739 | 3q22.2 | Аутосомно-рецессивный | Младенческий | ||
| SPG? | PLA2G6 | 22q13.1 | Аутосомно-рецессивный | Чайлд ood | ||
| SPG? | 1p36.33 | Аутосомно-доминантный | Детство | |||
| SPG? | KCNA2 | 1p13.3 | Аутосомно-доминантный | Детство | ||
| SPG? | Гранулин | 17q21.31 | ||||
| SPG? | POLR3A | 10q22.3 | Аутосомно-рецессивный |
Основным признаком HSP является зависимая от длины дегенерация аксонов. К ним относятся перекрещенные и неперекрещенные кортикоспинальные пути к ногам и fasciculus gracilis. Спиноцеребеллярный тракт задействован в меньшей степени. Тела нервных клеток дегенерирующих аксонов сохранены, и нет никаких доказательств первичной демиелинизации. В некоторых случаях наблюдается выпадение клеток переднего рога спинного мозга. Ганглии задних корешков, задние корешки и периферические нервы не затрагиваются напрямую.
HSP влияет на несколько путей в двигательных нейронах. Многие гены были идентифицированы и связаны с HSP. Остается проблема точно определить ключевых игроков в каждом из затронутых путей, в основном потому, что многие гены выполняют несколько функций и участвуют более чем в одном пути (рисунок).
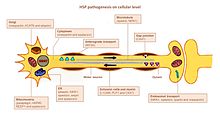 Обзор патогенеза HSP на клеточном уровне. Изображены идентифицированные затронутые гены в каждом пути.
Обзор патогенеза HSP на клеточном уровне. Изображены идентифицированные затронутые гены в каждом пути. Поиск пути важен для роста аксона в нужном направлении (например, в другую нервную клетку или мышцу). Важным для этого механизма является ген L1CAM, гликопротеин клеточной поверхности суперсемейства иммуноглобулинов. Мутации, ведущие к потере функции в L1CAM, также обнаруживаются при других Х-сцепленных синдромах. Все эти расстройства демонстрируют поражение кортикоспинального тракта (отличительный признак HSP). L1CAM участвует в наборе взаимодействий, связывая другие молекулы L1CAM, а также молекулы адгезии внеклеточных клеток, интегрины и протеогликаны или внутриклеточные белки, такие как анкирины.
Дефект поиска пути возникает из-за ассоциации L1CAM с нейропилином. -1. Нейропилин-1 взаимодействует с белками плексин-А с образованием рецепторного комплекса семафорин-3А. Затем семафорин-a3A высвобождается в вентральном отделе спинного мозга, чтобы отвести кортикоспинальные нейроны от срединного соединения спинного мозга / костного мозга. Если L1CAM не работает правильно из-за мутации, кортиокоспинальные нейроны не направляются в правильное положение, и возникает нарушение.
Аксоны в центральной и периферической нервной системе покрыты оболочкой с изоляцией, слоем миелина, для увеличения скорости распространения потенциала действия. Аномальная миелинизация в ЦНС обнаруживается при некоторых формах HSP HSP. Несколько генов были связаны с мальформацией миелина, а именно PLP1, GFC2 и FA2H. Мутации изменяют состав, толщину и целостность миелина.
Эндоплазматический ретикулум (ER) является основной органеллой для синтеза липидов. Мутации в генах, кодирующих белки, которые играют роль в формировании морфологии ER и метаболизма липидов, были связаны с HSP. Мутации в ATL1, BSCL2 и ERLIN2 изменяют структуру ER, в частности сеть канальцев и образование трехсторонних соединений в канальцах ER. Многие мутировавшие гены связаны с аномальным метаболизмом липидов. Наиболее распространено влияние на метаболизм арахидоновой кислоты (CYP2U1 ) и холестерина (CYP7B1 ), активность фосфолипазы (DDHD1 и DDHD2), образование ганглиозидов (B4GALNT-1 ) и баланс между углеводным и жировым обменом (SLV33A1).
Нейроны поглощают вещества из окружающей среды посредством эндоцитоза. Эндоцитарные везикулы сливаются с эндосомами, чтобы высвободить их содержимое. Транспортировка эндосом осуществляется по трем основным компартментам: Гольджи в / из эндосом; плазматическая мембрана в / из ранних эндосом (через рециклинг эндосом) и поздних эндосом в лизосомы. Как сообщается в HSP, дисфункция эндосомного транспорта может иметь серьезные последствия для мотонейронов с длинными аксонами. Мутации в AP4B1 и KIAA0415 связаны с нарушением образования везикул и мембранного транспорта, включая избирательное поглощение белков везикулами. Оба гена кодируют белки, которые взаимодействуют с несколькими другими белками и нарушают секреторные и эндоцитарные пути.
Дисфункции митохондрий связаны с онтогенетическими и дегенеративными неврологическими расстройствами. Только несколько генов HSP кодируют митохондриальные белки. Два митохондриальных резидентных белка мутировали в HSP: параплегин и шаперонин 60. Параплегин представляет собой металлопротеазу m-AAA внутренней митохондриальной мембраны. Он участвует в сборке рибосом и контроле качества белка. Нарушение активности шаперонина 60 приводит к нарушению контроля качества митохондрий. Два гена DDHD1 и CYP2U1 показали изменение митохондриальной архитектуры в фибробластах пациента. Эти гены кодируют ферменты, участвующие в метаболизме жирных кислот.
Первоначальная диагностика HSP основана на семейном анамнезе, наличии или отсутствии дополнительных признаков и исключении других негенетических причин спастичности, последнее особенно важно в спорадических случаях.
Церебральный и спинной МРТ - важная процедура, выполняемая для исключения других частых неврологических состояний, таких как рассеянный склероз, но также для выявления связанных аномалий, таких как атрофия мозжечка или мозолистого тела, а также аномалии белого вещества. Дифференциальный диагноз HSP должен также исключать спастическую диплегию, которая проявляется почти с одинаковыми повседневными эффектами и даже поддается лечению аналогичными лекарствами, такими как баклофен и ортопедическая хирургия ; время от времени эти два состояния могут выглядеть и ощущаться настолько похожими, что единственное различие может заключаться в наследственной природе HSP по сравнению с явно ненаследственной природой спастической диплегии (однако, в отличие от спастической диплегии и других форм спастического церебрального паралича, HSP нельзя надежно лечить с помощью селективной дорсальной ризотомии ).
Окончательное подтверждение диагноза HSP может быть предоставлено только путем проведения генетических тестов, направленных на известные генетические мутации.
Наследственные спастические параплегии могут быть классифицированы на основании симптомов; режим наследования; возраст пациента в начале заболевания; пораженные гены; и задействованные биохимические пути.
Неизвестно специфическое лечение, которое могло бы предотвратить, замедлить или обратить вспять HSP. Доступные методы лечения в основном состоят из симптоматического лечения и улучшения физического и эмоционального благополучия. Терапевтические препараты, предлагаемые пациентам с HSP, включают:
Хотя HSP является прогрессирующим заболеванием, прогноз для людей с HSP сильно различается. В первую очередь это поражает ноги, хотя у некоторых людей может быть поражение верхней части тела. В одних случаях это приводит к серьезной инвалидности, в то время как в других - меньше, и они совместимы с продуктивной и полноценной жизнью. Большинство людей с HSP имеют нормальную ожидаемую продолжительность жизни.
По оценкам, во всем мире распространенность всех наследственных спастических параплегий вместе взятых составляет от 2 до 6 на 100 000 человек. Норвежское исследование более 2,5 миллионов человек, опубликованное в марте 2009 г., показало, что уровень распространенности HSP составляет 7,4 на 100 000 населения - более высокий показатель, но в том же диапазоне, что и в предыдущих исследованиях. Никаких различий в частоте, связанных с полом, обнаружено не было, средний возраст начала заболевания составлял 24 года. В США наследственная спастическая параплегия внесена в список «редких заболеваний» Управлением по редким заболеваниям (ORD) Национальных институтов здравоохранения, что означает, что заболевание затрагивает меньше более 200000 человек в населении США.
| Классификация | D |
|---|---|
| Внешние ресурсы |