
Войти
ChIP-секвенирование, также известное как ChIP-seq - это метод, используемый для анализа взаимодействий белка с ДНК. ChIP-seq объединяет иммунопреципитацию хроматина (ChIP) с массивно параллельным секвенированием ДНК для идентификации сайтов связывания ДНК-ассоциированных белков. Его можно использовать для точного картирования глобальных сайтов связывания для любого интересующего белка. Раньше ChIP-on-Chip был наиболее распространенной техникой, используемой для изучения этих отношений белок-ДНК.
ChIP-seq в основном используется для определения того, как факторы транскрипции и другие связанные с хроматином белки влияют на механизмы воздействия на фенотип. Определение того, как белки взаимодействуют с ДНК для регулирования экспрессии генов, необходимо для полного понимания многих биологических процессов и болезненных состояний. Эта эпигенетическая информация дополняет генотип и анализ экспрессии. Технология ChIP-seq в настоящее время рассматривается в первую очередь как альтернатива ChIP-chip, которая требует гибридизационного массива . Это вносит некоторую предвзятость, так как массив ограничен фиксированным числом датчиков. Секвенирование, напротив, считается менее систематическим, хотя систематическая ошибка различных технологий секвенирования еще не полностью изучена.
Конкретные участки ДНК, находящиеся в прямом физическом взаимодействии с факторами транскрипции и другими белками, могут быть выделены с помощью иммунопреципитации хроматина. ChIP производит библиотеку сайтов-мишеней ДНК, связанных с интересующим белком in vivo. Анализы массово параллельных последовательностей используются в сочетании с базами данных полногеномных последовательностей для анализа характера взаимодействия любого белка с ДНК или характера любых эпигенетических модификаций хроматина. Это может быть применено к набору ChIP-способных белков и модификаций, таких как факторы транскрипции, полимеразы и механизмы транскрипции, структурные белки, белок. модификации, и. В качестве альтернативы зависимости от специфических антител были разработаны различные методы для поиска расширенного набора всех нуклеосомных -деплетированных или поврежденных нуклеосомами активных регуляторных областей в геноме, например DNase-Seq и FAIRE-Seq.
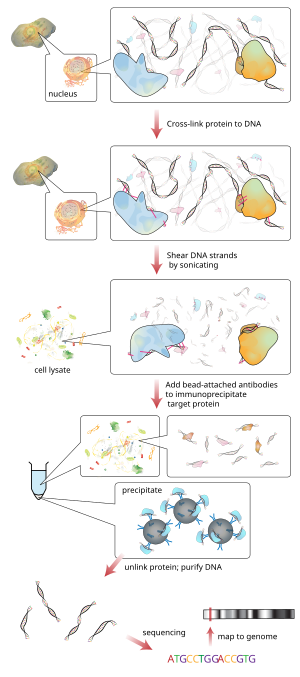 Рабочий процесс ChIP-секвенирования
Рабочий процесс ChIP-секвенирования ChIP - мощный метод выборочного обогащения последовательностей ДНК, связанных с конкретным белком в живые клетки. Однако широкое использование этого метода ограничивается отсутствием достаточно надежного метода для идентификации всех обогащенных последовательностей ДНК. Протокол влажной лаборатории ChIP содержит ChIP и гибридизацию. По сути, протокол ChIP состоит из пяти частей, которые помогают лучше понять общий процесс ChIP. Чтобы выполнить ChIP, первым шагом является перекрестное связывание с использованием формальдегида и больших партий ДНК для получения полезного количества. Поперечные связи осуществляются между белком и ДНК, а также между РНК и другими белками. Второй шаг - это процесс фрагментации хроматина, который разбивает хроматин, чтобы в конечном итоге получить высококачественные фрагменты ДНК для анализа ChIP. Эти фрагменты следует разрезать так, чтобы каждый из них составлял менее 500 пар оснований, чтобы получить наилучший результат для картирования генома. Третий этап называется иммунопреципитацией хроматина, сокращенно от ChIP. Процесс ChIP усиливает специфические сшитые комплексы ДНК-белок с использованием антитела против представляющего интерес белка с последующей инкубацией и центрифугированием для получения иммунопреципитации. Стадия иммунопреципитации также позволяет удалить неспецифические сайты связывания. Четвертый этап - это восстановление и очистка ДНК, происходящая путем обратного воздействия на перекрестные связи между ДНК и белком, чтобы разделить их и очистить ДНК экстракцией. Пятый и последний этап - это этап анализа протокола ChIP посредством процесса qPCR, ChIP-on-chip (гибридный массив) или секвенирования ChIP. Олигонуклеотидные адаптеры затем добавляются к небольшим участкам ДНК, которые были связаны с интересующим белком, чтобы обеспечить массовое параллельное секвенирование. Затем с помощью анализа последовательности могут быть идентифицированы и интерпретированы геном или областью, с которой был связан белок.
После выбора размера все полученные фрагменты ChIP-ДНК секвенируются одновременно с использованием секвенатора генома. За один прогон секвенирования можно сканировать ассоциации по всему геному с высоким разрешением, что означает, что особенности могут быть расположены точно на хромосомах. ChIP-чип, напротив, требует больших наборов тайлинговых массивов для более низкого разрешения.
На этом этапе секвенирования используется много новых методов секвенирования. Некоторые технологии, которые анализируют последовательности, могут использовать прикрепленные адаптером фрагменты ДНК ChIP на твердом субстрате для создания кластеров примерно по 1000 клональных копий каждый. Получающийся в результате массив кластеров матрицы с высокой плотностью на поверхности проточной кюветы секвенируется программой анализа генома. Каждый матричный кластер подвергается секвенированию путем синтеза параллельно с использованием новых флуоресцентно меченных нуклеотидов с обратимым терминатором. Шаблоны упорядочиваются по основанию во время каждого чтения. Затем программное обеспечение для сбора и анализа данных сопоставляет последовательности образцов с известной геномной последовательностью для идентификации фрагментов ChIP-ДНК.
Чувствительность этой технологии зависит от глубины цикла секвенирования ( т. е. количество отображаемых тегов последовательностей), размер генома и распределение целевого фактора. Глубина секвенирования напрямую связана со стоимостью. Если многочисленные связывающие вещества в больших геномах необходимо картировать с высокой чувствительностью, затраты высоки, поскольку потребуется чрезвычайно большое количество тегов последовательности. В этом отличие от ChIP-чипа, в котором стоимость не коррелирует с чувствительностью.
В отличие от методов ChIP на основе микроматрицы, точность анализа ChIP-seq не ограничивается расстоянием между заранее определенными зондами. За счет интеграции большого количества коротких считываний достигается высокоточная локализация сайта связывания. По сравнению с ChIP-чипом, данные ChIP-seq можно использовать для определения местоположения сайта связывания в пределах нескольких десятков пар оснований от фактического сайта связывания белка. Плотность меток в сайтах связывания является хорошим индикатором аффинности связывания белок-ДНК, что упрощает количественную оценку и сравнение аффинности связывания белка с разными сайтами ДНК.
ДНК-ассоциация STAT1: ChIP-seq использовали для исследования мишеней STAT1 в клетках HeLA S3, которые являются клонами линии HeLA, которые используются для анализа клеточных популяций. Затем эффективность ChIP-seq сравнивали с альтернативными методами взаимодействия белок-ДНК, такими как ChIP-PCR и ChIP-chip.
Архитектура промоторов нуклеосом: Используя ChIP-seq, было определено, что дрожжевые гены, по-видимому, могут имеют минимальную свободную от нуклеосом промоторную область размером 150 п.н., в которой РНК-полимераза может инициировать транскрипцию.
Сохранение фактора транскрипции: ChIP-seq использовали для сравнения консервативности TF в переднем мозге и ткани сердца у эмбриональных мышей. Авторы определили и подтвердили функциональность сердца энхансеров транскрипции и определили, что энхансеры транскрипции для сердца менее консервативны, чем энхансеры для переднего мозга, на той же стадии развития.
ChIP-seq по всему геному: ChIP-секвенирование было завершено на черве C. elegans для исследования сайтов связывания 22 транскрипционных факторов по всему геному. До 20% аннотированных генов-кандидатов были отнесены к факторам транскрипции. Несколько факторов транскрипции были отнесены к некодирующим областям РНК и могут зависеть от факторов развития или окружающей среды. Также были определены функции некоторых факторов транскрипции. Некоторые факторы транскрипции регулируют гены, которые контролируют другие факторы транскрипции. Эти гены не регулируются другими факторами. Большинство факторов транскрипции служат как мишенями, так и регуляторами других факторов, демонстрируя сеть регуляции.
Предполагаемая регуляторная сеть: сигнал ChIP-seq модификации гистона в большей степени коррелирует с транскрипцией. факторные мотивы на промоторах по сравнению с уровнем РНК. Следовательно, автор предположил, что использование модификации гистонов ChIP-seq обеспечит более надежный вывод о ген-регуляторных сетях по сравнению с другими методами, основанными на экспрессии.
ChIP-seq предлагает альтернативу ChIP-chip. Экспериментальные данные ChIP-seq STAT1 имеют высокую степень сходства с результатами, полученными с помощью ChIP-chip для того же типа эксперимента, с более чем 64% пиков в общих геномных областях. Поскольку данные представляют собой считываемые последовательности, ChIP-seq предлагает конвейер быстрого анализа, если для картирования считывания доступна высококачественная последовательность генома и геном не имеет повторяющегося содержимого, которое затрудняет процесс картирования. ChIP-seq также может обнаруживать мутации в последовательностях сайтов связывания, которые могут напрямую поддерживать любые наблюдаемые изменения в связывании белков и регуляции генов.
Как и многие подходы к высокопроизводительному секвенированию, ChIP-seq генерирует чрезвычайно большие наборы данных, для которых требуются соответствующие методы вычислительного анализа. Для прогнозирования сайтов связывания ДНК по данным подсчета считывания ChIP-seq были разработаны методы вызова пика. Самый популярный метод - MACS, который эмпирически моделирует размер сдвига тегов ChIP-Seq и использует его для улучшения пространственного разрешения прогнозируемых сайтов привязки.
Другой актуальной вычислительной проблемой является дифференциальный пиковый вызов, который выявляет значительные различия в двух сигналах ChIP-seq из разных биологических условий. Вызывающие дифференциальные пики сегментируют два сигнала ChIP-seq и идентифицируют дифференциальные пики с помощью скрытых марковских моделей. Примерами двухступенчатых дифференциальных пиков, вызывающих вызовы, являются ChIPDiff и ODIN.