
Войти
A геномная библиотека - это совокупность полной геномной ДНК одного организма. ДНК хранится в популяции идентичных векторов, каждый из которых содержит различную вставку ДНК. Для создания геномной библиотеки ДНК организма экстрагируют из клеток, а затем переваривают рестрикционным ферментом для разрезания ДНК на фрагменты определенного размера.. Затем фрагменты вставляют в вектор с помощью ДНК-лигазы. Затем векторная ДНК может быть поглощена организмом-хозяином - обычно популяцией Escherichia coli или дрожжей - с каждой клеткой, содержащей только одну векторную молекулу. Использование клетки-хозяина для переноса вектора позволяет легко амплификацию и извлечение конкретных клонов из библиотеки для анализа.
Есть несколько доступны виды векторов с различной емкостью вставки. Как правило, библиотеки, сделанные из организмов с более крупными геномами, требуют векторов с более крупными вставками, поэтому для создания библиотеки требуется меньше векторных молекул. Исследователи могут выбрать вектор, также учитывая идеальный размер вставки, чтобы найти желаемое количество клонов, необходимых для полного покрытия генома.
Геномные библиотеки обычно используются для приложений секвенирования. Они сыграли важную роль в секвенировании всего генома нескольких организмов, включая геном человека и несколько модельных организмов.
Первый полностью секвенированный геном на основе ДНК был создан двукратным лауреатом Нобелевской премии Фредерик Сэнгер, в 1977 году. Сэнгер и его команда ученых создали библиотеку бактериофага, phi X 174 для использования в ДНК sequ. применение. Важность этого успеха способствовала постоянно растущему спросу на секвенирование геномов для исследования генной терапии. Теперь команды могут каталогизировать полиморфизмы в геномах и исследовать те гены-кандидаты, которые вносят вклад в такие заболевания, как болезнь Паркинсона, болезнь Альцгеймера, рассеянный склероз, ревматоидный артрит и диабет 1 типа. Это связано с продвижением полногеномных ассоциативных исследований с возможностью создания и секвенирования геномных библиотек. Ранее одними из подходов были исследования сцепления и генов-кандидатов.
Конструирование геномной библиотеки включает создание множества рекомбинантных молекул ДНК. Геномную ДНК организма экстрагируют, а затем расщепляют с помощью рестрикционного фермента. Для организмов с очень маленькими геномами (~ 10 т.п.н.) расщепленные фрагменты могут быть разделены с помощью гель-электрофореза. Затем разделенные фрагменты можно вырезать и клонировать в вектор отдельно. Однако, когда большой геном переваривается рестрикционным ферментом, остается слишком много фрагментов, чтобы вырезать их по отдельности. Весь набор фрагментов должен быть клонирован вместе с вектором, после чего может произойти разделение клонов. В любом случае фрагменты лигируют в вектор, который был переварен тем же рестрикционным ферментом. Затем вектор, содержащий вставленные фрагменты геномной ДНК, может быть введен в организм-хозяин.
Ниже приведены этапы создания геномной библиотеки из большого генома.
Ниже приведена диаграмма вышеизложенного. шаги.
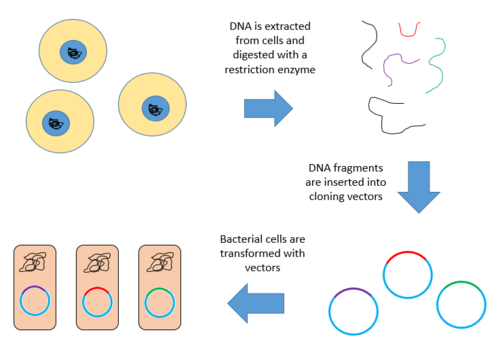 Конструирование геномной библиотеки
Конструирование геномной библиотеки После конструирования геномной библиотеки с использованием вирусного вектора, такого как фаг лямбда, титр библиотеку можно определить. Расчет титра позволяет исследователям приблизительно определить, сколько инфекционных вирусных частиц было успешно создано в библиотеке. Для этого используются разведения библиотеки для трансформации культур E.coli известных концентраций. Затем культуры высевают на чашки с агаром и инкубируют в течение ночи. Подсчитывается количество вирусных бляшек, и их можно использовать для расчета общего количества инфекционных вирусных частиц в библиотеке. Большинство вирусных векторов также несут маркер, который позволяет отличать клоны, содержащие вставку, от тех, которые не имеют вставки. Это позволяет исследователям также определять процент инфекционных вирусных частиц, фактически несущих фрагмент библиотеки.
Аналогичный метод можно использовать для титрования геномных библиотек, созданных с помощью невирусных векторов, таких как плазмиды и BAC. Тест лигирования библиотеки можно использовать для трансформации E. coli. Затем трансформацию наносят на чашки с агаром и инкубируют в течение ночи. Титр трансформации определяют путем подсчета количества колоний, присутствующих на чашках. Эти векторы обычно имеют селективный маркер , позволяющий дифференцировать клоны, содержащие вставку, от клонов, которые не содержат. Выполняя этот тест, исследователи также могут определить эффективность лигирования и внести необходимые корректировки, чтобы обеспечить получение желаемого количества клонов для библиотеки.
 Гибридизация колоний блотом
Гибридизация колоний блотом В Чтобы выделить клоны, содержащие интересующие области, из библиотеки, библиотека должна быть сначала проверена. Одним из методов скрининга является гибридизация. Каждая трансформированная клетка-хозяин библиотеки будет содержать только один вектор с одной вставкой ДНК. Всю библиотеку можно поместить на фильтр поверх носителя. Фильтр и колонии готовят для гибридизации и затем метят зондом . Целевая ДНК-вставка, представляющая интерес, может быть идентифицирована путем обнаружения, такого как авторадиография, из-за гибридизации с зондом, как показано ниже.
Другой метод скрининга - это полимеразная цепная реакция (ПЦР). Некоторые библиотеки хранятся в виде пулов клонов, и скрининг с помощью ПЦР является эффективным способом идентификации пулов, содержащих определенные клоны.
Размер генома варьируется у разных организмов, и клонирование вектор должен быть выбран соответственно. Для большого генома следует выбирать вектор с большой емкостью, чтобы относительно небольшое количество клонов было достаточным для покрытия всего генома. Однако часто бывает труднее охарактеризовать вставку, содержащуюся в векторе с большей емкостью.
Ниже представлена таблица нескольких типов векторов, обычно используемых для геномных библиотек, и размера вставки, которые каждая в целом держится.
| Вектор тип | Вставка размер (в тысячах) |
|---|---|
| Плазмиды | до 10 |
| Лямбда фага (λ) | до 25 |
| Космиды | до 45 |
| Бактериофаг P1 | от 70 до 100 |
| Искусственные хромосомы P1 (PACs) | 130 до 150 |
| Бактериальные искусственные хромосомы (BACs) | от 120 до 300 |
| Дрожжевые искусственные хромосомы (YAC) | от 250 до 2000 |
A плазмида представляет собой двухцепочечную кольцевую ДНК молекула, обычно используемая для молекулярного клонирования. Плазмиды обычно имеют длину от 2 до 4 (т.п.н.) и способны нести вставки размером до 15 т.п.н. Плазмиды содержат точку начала репликации, позволяющую им реплицироваться внутри бактерии независимо от хромосомы хозяина. Плазмиды обычно несут ген устойчивости к антибиотикам, который позволяет отобрать бактериальные клетки, содержащие плазмиду. Многие плазмиды также несут репортерный ген, который позволяет исследователям отличать клоны, содержащие вставку, от тех, которые ее не содержат.
Фаг λ является вирус с двухцепочечной ДНК, инфицирующий E. coli. Λ-хромосома имеет длину 48,5 КБ и может содержать вставки размером до 25 КБ. Эти вставки заменяют несущественные вирусные последовательности в λ-хромосоме, в то время как гены, необходимые для образования вирусных частиц и инфекции, остаются нетронутыми. ДНК вставки реплицируется с вирусной ДНК; таким образом, вместе они упакованы в вирусные частицы. Эти частицы очень эффективны при заражении и размножении, что приводит к более высокому производству рекомбинантных λ-хромосом. Однако из-за меньшего размера вставки библиотеки, созданные с фагом λ, могут потребовать много клонов для полного покрытия генома.
Космиды векторы - это плазмиды, которые содержат небольшую область ДНК бактериофага λ, называемую последовательность cos. Эта последовательность позволяет упаковывать космиду в частицы бактериофага λ. Эти частицы, содержащие линеаризованную космиду, вводятся в клетку-хозяина посредством трансдукции. Попав внутрь хозяина, космиды циркулируют с помощью ДНК-лигазы хозяина и затем функционируют как плазмиды. Космиды способны нести вставки размером до 40 килобайт.
векторы бактериофага P1 могут содержать вставки размером 70-100 килобайт. Они начинаются как линейные молекулы ДНК, упакованные в частицы бактериофага P1. Эти частицы вводят в штамм E. coli, экспрессирующий Cre-рекомбиназу. Линейный вектор P1 становится кольцевым за счет рекомбинации между двумя сайтами loxP в векторе. Векторы P1 обычно содержат ген устойчивости к антибиотикам и маркер положительной селекции, позволяющий отличать клоны, содержащие вставку, от клонов, которые не содержат. Векторы P1 также содержат плазмиду P1 репликон, которая гарантирует, что в клетке присутствует только одна копия вектора. Однако существует второй репликон P1, называемый литическим репликоном P1, который контролируется индуцибельным промотором. Этот промотор позволяет амплифицировать более одной копии вектора на клетку перед экстракцией ДНК.
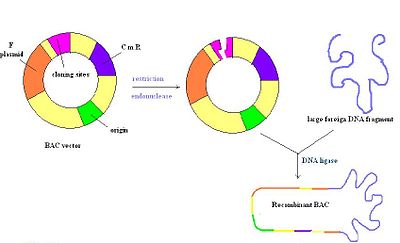 вектор bac
вектор bac искусственные хромосомы P1 (PAC) имеют особенности обоих P1 векторы и бактериальные искусственные хромосомы (BAC). Подобно векторам P1, они содержат плазмиду и литический репликон, как описано выше. В отличие от векторов P1, их не нужно упаковывать в частицы бактериофага для трансдукции. Вместо этого они вводятся в E. coli в виде кольцевых молекул ДНК посредством электропорации, как и ВАС. Также как и ВАС, их относительно сложнее приготовить из-за единственного источника репликации.
Бактериальные искусственные хромосомы (ВАС) представляют собой кольцевые молекулы ДНК, обычно длиной около 7 килобайт., которые могут содержать вставки размером до 300 КБ. Векторы ВАС содержат репликон, полученный из E. coli F-фактор, что обеспечивает сохранение одной копии на клетку. После лигирования вставки в ВАС, ВАС вводят в рекомбинационные штаммы E.coli путем электропорации. Большинство векторов ВАС содержат ген устойчивости к антибиотикам, а также маркер положительной селекции. На рисунке справа изображен вектор ВАС, разрезанный рестрикционным ферментом с последующей вставкой чужеродной ДНК, которая повторно отжигается лигазой. В целом, это очень стабильный вектор, но их может быть трудно приготовить из-за единственной точки начала репликации, как и у PAC.
Дрожжевые искусственные хромосомы (YAC) линейны Молекулы ДНК, содержащие необходимые признаки аутентичной хромосомы дрожжей, включая теломеры, центромеру и ориджин репликации. Большие вставки ДНК можно лигировать в середину YAC, так что есть «плечо» YAC с обеих сторон вставки. Рекомбинантный YAC вводят в дрожжи путем трансформации; выбираемые маркеры, присутствующие в YAC, позволяют идентифицировать успешные трансформанты. YAC могут содержать вставки размером до 2000 КБ, но большинство библиотек YAC содержат вставки размером 250-400 КБ. Теоретически не существует верхнего предела размера вставки, которую может вместить YAC. Именно качество подготовки ДНК, используемой для вставок, определяет предел размера. Наиболее сложным аспектом использования YAC является тот факт, что они подвержены перегруппировке.
Выбор вектора требует, чтобы созданная библиотека была репрезентативной для всего генома. Любая вставка генома, полученная из рестрикционного фермента, должна иметь равные шансы оказаться в библиотеке по сравнению с любой другой вставкой. Кроме того, рекомбинантные молекулы должны содержать достаточно большие вставки, обеспечивающие удобство работы с размером библиотеки. Это, в частности, определяется количеством клонов, которые необходимо иметь в библиотеке. Количество клонов для выборки всех генов определяется размером генома организма, а также средним размером вставки. Это представлено формулой (также известной как формула углерода и Кларка):
где,



где,


Таким образом, увеличивая размер вставки (путем выбора вектора) позволит уменьшить количество клонов, необходимых для представления генома. Отношение размера вставки к размеру генома представляет собой долю соответствующего генома в одном клоне. Вот уравнение со всеми рассматриваемыми частями:
Приведенная выше формула может использоваться для определения уровня достоверности 99% того, что все последовательности в геноме представлены с помощью вектора с вставить размер двадцати тысяч пар оснований (например, фаговый лямбда-вектор). Размер генома организма в этом примере составляет три миллиарда пар оснований.

Таким образом, примерно 688 060 клонов являются требуется для обеспечения 99% вероятности того, что данная последовательность ДНК из этого генома с тремя миллиардами пар оснований будет присутствовать в библиотеке с использованием вектора с размером вставки в двадцать тысяч пар оснований.
После создания библиотеки геном организма можно секвенировать, чтобы выяснить, как гены влияют на организм, или сравнить похожие организмы на уровне генома. Вышеупомянутые общегеномные исследования ассоциации могут идентифицировать гены-кандидаты, происходящие от многих функциональных признаков. Гены можно выделить с помощью геномных библиотек и использовать на линиях клеток человека или животных моделях для дальнейших исследований. Кроме того, создание клонов с высокой точностью воспроизведения с точным представлением генома и без проблем со стабильностью может также стать промежуточным звеном для секвенирования с дробовиком или изучения полных генов в функциональном анализе.
 Полногеномное секвенирование по сравнению с иерархическим секвенированием
Полногеномное секвенирование по сравнению с иерархическим секвенированием Одним из основных видов использования геномных библиотек является иерархическое секвенирование с дробовиком, которое также называется нисходящим секвенированием, на основе карты или клон-за-клоном. Эта стратегия была разработана в 1980-х годах для секвенирования целых геномов до того, как стали доступны высокопроизводительные методы секвенирования. Отдельные клоны из геномных библиотек можно разрезать на более мелкие фрагменты, обычно от 500 до 1000 пар оснований, которые более удобны для секвенирования. После секвенирования клона из геномной библиотеки последовательность можно использовать для скрининга библиотеки на наличие других клонов, содержащих вставки, которые перекрываются с секвенированным клоном. Затем можно секвенировать любые новые перекрывающиеся клоны с образованием контига . Этот метод, называемый ходьбой по хромосомам, может использоваться для секвенирования целых хромосом.
Секвенирование всего генома - еще один метод секвенирования генома, который не требует библиотеки векторов высокой емкости. Скорее, он использует компьютерные алгоритмы для сборки коротких считываний последовательностей, охватывающих весь геном. По этой причине геномные библиотеки часто используются в сочетании с секвенированием всего генома. Карта высокого разрешения может быть создана путем секвенирования обоих концов вставок из нескольких клонов в геномной библиотеке. Эта карта предоставляет последовательности известных расстояний друг от друга, которые могут использоваться для помощи в сборке считываний последовательностей, полученных с помощью секвенирования дробовика. Последовательность генома человека, которая была объявлена завершенной в 2003 году, была собрана с использованием как библиотеки ВАС, так и секвенирования дробовика.
Исследования ассоциаций всего генома являются общими приложениями для поиска специфические генные мишени и полиморфизмы в пределах человеческого рода. Фактически, международный проект HapMap был создан благодаря партнерству ученых и агентств из нескольких стран для каталогизации и использования этих данных. Цель этого проекта - сравнить генетические последовательности разных людей, чтобы выявить сходства и различия в хромосомных областях. Ученые из всех участвующих стран каталогизируют эти атрибуты с данными о популяциях африканского, азиатского и европейского происхождения. Такие общегеномные оценки могут привести к дальнейшим диагностическим и медикаментозным методам лечения, а также помочь будущим командам сосредоточиться на организации терапии с учетом генетических особенностей. Эти концепции уже используются в генной инженерии. Например, исследовательская группа фактически сконструировала челночный вектор PAC, который создает библиотеку, представляющую двукратный охват генома человека. Это может послужить невероятным ресурсом для идентификации генов или наборов генов, вызывающих болезнь. Более того, эти исследования могут служить мощным способом изучения регуляции транскрипции, как это было замечено при изучении бакуловирусов. В целом, успехи в создании библиотеки геномов и секвенировании ДНК позволили эффективно обнаруживать различные молекулярные мишени. Усвоение этих свойств с помощью таких эффективных методов может ускорить использование новых кандидатов в лекарственные препараты.
Klug, Cummings, Spencer, Palladino (2010). Основы генетики. Пирсон. С. 355–264. ISBN 978-0-321-61869-6. CS1 maint: несколько имен: список авторов (ссылка )



![N = {\ frac {ln (1-0.99)} {ln [1 - {\ frac {2.0 \ times 10 ^ {4} basepairs} {3.0 \ times 10 ^ {9} basepairs}}]}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/053b596ba7adfc402824b91ceae3dd24973b2fec)
