
Войти
| Синдром Крузона | |
|---|---|
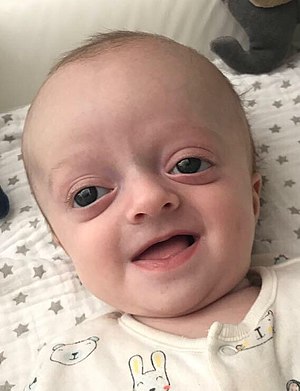 | |
| Ребенок с синдромом Крузона | |
| Специальность | Медицинская генетика |
Синдром Крузона - аутосомно-доминантное генетическое заболевание, известное как синдром жаберной дуги. В частности, этот синдром поражает первую жаберную (или глоточную) дугу, которая является предшественником верхней челюсти и нижней челюсти. Поскольку жаберные дуги являются важными элементами развития растущего эмбриона, нарушения в их развитии создают длительные и широко распространенные последствия.
Этот синдром назван в честь Октава Крузона, французского врача, который первым описал это заболевание. Сначала называется «черепно-лицевой дизостоз» («краниофациальный » относится к черепу и лицу, а «дизостоз » относится к порокам развития кости.) заболевание характеризовалось рядом клинических особенностей, которые можно описать рудиментарными значениями его прежнего названия. Этот синдром вызван мутацией рецептора 2 фактора роста фибробластов (FGFR2), расположенного на хромосоме 10. Череп и лицевые кости развивающегося плода рано срастаются или не могут расширяться. Таким образом, нормальный рост костей невозможен. Сращивание разных швов приводит к аномальному росту черепа.
 Ребенок с синдромом Крузона с характерными чертами лица.
Ребенок с синдромом Крузона с характерными чертами лица.  Черепные швы
Черепные швы Определяющей характеристикой синдрома Крузона является краниосиностоз, который приводит к аномалии головы форма. Это присутствует в комбинациях: туррицефалии, лобной выпуклости, тригоноцефалии (слияние метопического шва ), брахицефалии (сращение коронарного шва), долихоцефалия (слияние сагиттального шва ), плагиоцефалия (одностороннее преждевременное ушивание лямбдоида и коронковые швы ), оксицефалия (слияние коронарных и лямбдоидных швов ) и сложный краниосиностоз (преждевременный закрытие некоторых или всех швов).
экзофтальм (выпуклые глаза из-за неглубоких глазниц после раннего сращения окружающих костей), гипертелоризм (расстояние между глазами больше нормы) и (клюв- как нос) тоже очень общие черты. Другие черты лица, которые присутствуют во многих случаях, включают внешнее косоглазие и гипоплазию верхней челюсти (недостаточный рост средней зоны лица), что приводит к относительному прогнатизму нижней челюсти (выступающий подбородок) и дает эффект вогнутого лица пациента.
Большинство симптомов вторичны по отношению к аномальному строению черепа. Примерно у 30% людей с синдромом Крузона развивается гидроцефалия. Нейросенсорная тугоухость присутствует в некоторых случаях. Аномалии расположения глаз в глазницах могут вызывать проблемы со зрением, наиболее распространенным из которых является обнажение роговицы, которое может привести к ухудшению зрения. У некоторых людей с этим заболеванием ограничены дыхательные пути и могут возникнуть серьезные проблемы с дыханием.
Общие черты - узкое / высоко арочное небо, задний двусторонний перекрестный прикус, гиподонтия (отсутствие некоторых зубов), и скученность зубов. Из-за гипоплазии верхней челюсти люди с синдромом Крузона обычно имеют значительный постоянный прикус.
Текущее исследование указывает на рецепторы фактора роста фибробластов (FGFR) FGFR2 и FGFR3 как ведущие факторы, вызывающие аутосомно-доминантный синдром Крузона. Эти два трансмембранных белка являются двумя из четырех рецепторов фактора роста фибробластов, участвующих в дифференцировке остеобластов во время эмбрионального развития ; мутации среди этих рецепторов участвуют в нескольких генетических нарушениях. Известно 40 мутаций, большинство из которых вызвано миссенс-мутацией. FGFR2 является наиболее часто мутированным геном, отсутствующим в цистеине 342 в экзоне 9, который создает усиление функции. FGFR2lllc изоформа, созданная посредством альтернативного сплайсинга экзона 3 гена FGFR2, использует экзон 9 и используется в мезенхимальных стволовых клетках для контроля оссификации. Однако мутация постоянно активирует трансмембранный белок через дисульфидную связь, образованную неправильно из-за потери цистеина 342. FGFR3 экспрессируется больше в лобных костях во время эмбриональное развитие, направляющее развитие черепной кости. Точечная мутация вызывает конститутивную активацию тирозина в петле активации, расположенной в цитозольной области белка, что приводит к ускоренной дифференцировке лобных остеобластов, что приводит к преждевременному слиянию лобных костей черепа..
Диагноз синдрома Крузона обычно может быть поставлен при рождении путем оценки внешнего вида младенца. Дальнейший анализ, включая рентгенограммы, магнитно-резонансную томографию (МРТ), генетическое тестирование, рентген и компьютерную томографию, может быть использован для подтверждения диагноза синдрома Крузона.
 Аномальное сращение костей черепа характерно для синдрома Крузона.
Аномальное сращение костей черепа характерно для синдрома Крузона. Хирургия обычно используется для предотвращения закрытия швов черепа от повреждения развития мозга. Без хирургического вмешательства типичными исходами являются слепота и умственная отсталость. Чтобы переместить орбиты вперед, хирурги обнажают череп и орбиты и изменяют форму кости. Чтобы вылечить недостаточность средней зоны лица, хирурги могут переместить вперед нижнюю орбиту и кости средней зоны лица.
Людям с синдромом Крузона обычно накладывают несколько швов, в частности двусторонний коронарный краниосиностоз, и либо открытый свод. Может быть проведена операция или полосатая трепанация черепа (если ребенку до 6 месяцев). В более позднем сценарии шлем носят в течение нескольких месяцев после операции.
После лечения аномалий свода черепа пациенты Крузона обычно продолжают жить нормальной жизнью.
Заболеваемость синдромом Крузона в настоящее время оценивается у 1,6 из каждых 100 000 человек и является наиболее распространенным синдромом краниостеноза.
Синдром Крузона впервые был описан Октавом Крузон в 1912 году. Он отметил, что больные были матерью и ее дочерью, что подразумевает генетическую основу.
| Классификация | D |
|---|---|
| Внешние ресурсы |
| Викискладе есть средства массовой информации, связанные с синдромом Крузона. |