
Войти
| Синдром Аперта | |
|---|---|
| Другие названия | Акроцефало-синдактилия 1 типа |
 | |
| Синдром Хэнд-ин-Аперта с синдактилия | |
| Специальность | Медицинская генетика |
Синдром Аперта - это форма акроцефалосиндактилии, врожденного заболевания, характеризующегося пороками развития черепа, лица, руки и ноги. Он классифицируется как синдром жаберной дуги, поражающий первую жаберную (или глоточную) дугу, предшественник верхней челюсти и нижней челюсти. Нарушения развития жаберных дуг в процессе развития плода оказывают стойкое и широко распространенное воздействие.
В 1906 году Эжен Аперт, французский врач, описал девять человек, обладающих схожими атрибутами и характеристиками. С лингвистической точки зрения в термине «акроцефалосиндактилия» acro - это греческое для «пика», относящегося к «остроконечной» голове, которая часто встречается при синдроме; cephalo, также из греческого, - объединяющая форма, означающая «голова»; синдактилия относится к перепонкам пальцев рук и ног.
В эмбриологии руки и ноги имеют селективные клетки, которые умирают в процессе, называемом селективной смертью клеток, или апоптозом, вызывая разделение пальцев. В случае акроцефалосиндактилии селективной гибели клеток не происходит, и кожа, а в редких случаях кости между пальцами рук и ног срастается.
Также поражаются кости черепа, аналогично синдрому Крузона и синдрому Пфайффера. Краниосиностоз возникает, когда череп плода и лицевые кости сливаются слишком рано в утробе, нарушая нормальный рост костей. Слияние разных швов приводит к разным формам роста на черепе. Примеры включают: тригоноцефалию (слияние метопической нити ), брахицефалию (слияние коронарного шва и лямбдовидного шва с двух сторон), долихоцефалия (слияние сагиттального шва ), плагиоцефалия (слияние венечных и лямбдоидальных швов в одностороннем порядке) и оксицефалия или туррицефалия (слияние венечных и лямбдовидных швов).
Данные о заболеваемости синдромом в популяции различались: по оценкам, всего 1 из 200 000 рождений и 160 000 в среднем по более ранним исследованиям. Однако исследование, проведенное в 1997 году Калифорнийской программой мониторинга врожденных дефектов, показало, что уровень заболеваемости составляет 1 на 80 645 из почти 2,5 миллиона живорождений. Другое исследование, проведенное в 2002 году Черепно-лицевым центром Детской больницы Северного Техаса, показало более высокую заболеваемость - примерно 1 на 65 000 живорождений.
Черепные аномалии - наиболее очевидные последствия акроцефалосиндактилии. Краниосиностоз возникает, когда черепные швы закрываются слишком рано, хотя мозг ребенка все еще растет и расширяется. Брахицефалия - это распространенная модель роста, при которой коронковые швы закрываются преждевременно, предотвращая расширение черепа вперед или назад и заставляя мозг расширять череп в стороны и вверх. Это приводит к еще одной общей характеристике - высокому выступающему лбу с плоской задней частью черепа. Из-за преждевременного закрытия коронарных швов может развиться повышенное черепное давление, что приводит к умственной отсталости. Плоское или вогнутое лицо может развиться в результате недостаточного роста срединных костей лица, что приводит к состоянию, известному как псевдомандибулярный прогнатизм. Другие особенности акроцефалосиндактилии могут включать неглубокие костные орбиты и широко расставленные глаза. Низко посаженные уши также являются типичной характеристикой синдромов жаберных дуг.
Все синдромы акроцефалосиндактилии демонстрируют определенный уровень аномалий конечностей, поэтому их бывает сложно отличить друг от друга. Однако типичные деформации кисти у пациентов с синдромом Аперта отличают его от других синдромов. Руки у пациентов с синдромом Аперта всегда имеют четыре общих черты:
Деформация пространства между указательным и большим пальцами может быть переменной. Основываясь на этом первом веб-пространстве, мы можем различить три различных типа деформации руки:
| Тип I («лопата») | Тип II (« миттен ") | Тип III (" бутон розы ") | |
|---|---|---|---|
| Первое веб-пространство | Простая синдактилия | Простая синдактилия | Сложная синдактилия |
| Средняя тройка пальцы | Поперечное слияние с плоской ладонью | Слияние кончиков пальцев, образующих вогнутую ладонь | Плотное слияние всех пальцев одним соединенным ногтем |
| Четвертое перепончатое пространство | Простая и неполная синдактилия | Простая и полная синдактилия | Простая и полная синдактилия |
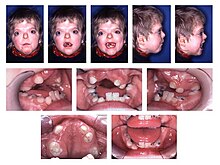 Зубы ребенка с синдромом Аперта
Зубы ребенка с синдромом Аперта Общие характерные особенности акроцефалосиндактилии - высокое арочное небо (выглядит как нижнечелюстной прогнатизм ), узкое небо и скученность зубов.
Омфалоцеле было описано у двух пациентов с синдромом Аперта Германом Т.Е. и другие. (США, 2010) и Ercoli G. et al. (Аргентина, 2014 г.). Омфалоцеле - это врожденный дефект, при котором кишечник или другие органы брюшной полости находятся вне тела младенца из-за отверстия в области пупка. Однако связь между омфалоцеле и синдромом Аперта еще не подтверждена, поэтому необходимы дополнительные исследования.
Акроцефалосиндактилия может быть аутосомно-доминантным расстройством. Мужчины и женщины страдают одинаково; однако исследования еще не установили точную причину. Тем не менее, почти все случаи носят спорадический характер, что свидетельствует о новых мутациях или экологическом воздействии на геном. Потомство родителей с синдромом Аперта имеет 50% шанс унаследовать это заболевание. В 1995 году A.O.M. Уилки опубликовал документ, демонстрирующий доказательства того, что акроцефалосиндактилия вызывается дефектом рецептора фактора роста фибробластов 2 гена, на хромосоме 10.
Синдром Аперта является аутосомно-доминантным заболеванием. ; приблизительно две трети случаев связаны с мутацией C в G в положении 755 гена FGFR2, которая вызывает изменение Ser в Trp в белке. Это горячая точка мужской мутации: в исследовании 57 случаев мутация всегда происходила на аллеле, полученном от отца. Исходя из наблюдаемой распространенности заболевания при рождении (1 из 70000), очевидная частота мутаций C в G на этом участке составляет около 0,00005, что в 200-800 раз выше, чем обычная частота мутаций в CG. динуклеотиды. Причем заболеваемость резко возрастает с возрастом отца. Goriely et al. (2003) проанализировали аллельное распределение мутаций в образцах спермы мужчин разного возраста и пришли к выводу, что простейшим объяснением данных является то, что мутация C в G дает клетке преимущество в мужской зародышевой линии.
Это До сих пор не очень ясно, почему у людей с синдромом Аперта наблюдается краниосиностоз и синдактилия. Одно исследование предполагает, что это имеет какое-то отношение к экспрессии трех изоформ FGFR2, гена с точечными мутациями, которые вызывают синдром у 98% пациентов. KGFR, рецептор фактора роста кератиноцитов, представляет собой изоформу, активную в метафизе и межфаланговых суставах. FGFR1 представляет собой изоформу, активную в диафизе. FGFR2-Bek активен как в метафизе, так и в диафизе, а также в межпальцевой мезенхиме. Точечная мутация увеличивает лиганд-зависимую активацию FGFR2 и, следовательно, его изоформ. Это означает, что FGFR2 теряет свою специфичность, вызывая связывание FGF, которые обычно не связываются с рецептором. Поскольку FGF подавляет апоптоз, межпальцевая мезенхима сохраняется. FGF также увеличивает репликацию и дифференцировку остеобластов, таким образом, раннее слияние нескольких швов черепа. Это может объяснить, почему при синдроме Аперта всегда обнаруживаются оба симптома.
Диагноз обычно ставится по очевидным физическим характеристикам и может быть подтвержден с помощью рентгеновского исследования черепа или компьютерной томографии головы. Молекулярное генетическое тестирование может подтвердить диагноз.
Хирургическое вмешательство необходимо для предотвращения закрытия венечных швов и нарушения развития мозга. В частности, операции по поводу или моноблочного остеогенеза средней зоны лица дистракционного остеогенеза, который отделяет среднюю часть лица или всю верхнюю часть лица, соответственно, от остальной части черепа, выполняются с целью репозиции их в правильной плоскости. Эти операции выполняются как пластическими, так и челюстно-лицевыми хирургами, часто в сотрудничестве.
Стандартного лечения пороков развития кисти в Apert не существует из-за различий и тяжести клинических проявлений у разных пациентов. Поэтому к каждому пациенту следует подходить и лечить индивидуально, стремясь к адекватному балансу между функциональностью руки и эстетикой. Однако в зависимости от степени деформации можно дать некоторые рекомендации. В общем, изначально рекомендуется освободить первое и четвертое межпальцевые промежутки, тем самым освободив пограничные лучи. Это позволяет ребенку брать предметы руками, что является очень важной функцией для его развития. Позже необходимо освободить второе и третье межпальцевые промежутки. Поскольку в Apert есть три типа рук, все со своими деформациями, все они нуждаются в разном подходе к их лечению:
По мере роста ребенка и, соответственно, рук, необходимы вторичные ревизии для лечения контрактур и улучшения эстетики.
| Классификация | D |
|---|---|
| Внешние ресурсы |