
Войти
Секвенирование микроРНК (miRNA-seq), тип RNA-Seq, используется секвенирования следующего поколения или массово-параллельного высокопроизводительного секвенирования ДНК с последовательностью микроРНК, также называемых миРНК. miRNA-seq отличается от других форм RNA-seq тем, что входной материал часто обогащен малыми РНК. miRNA-seq позволяет исследователям изучать тканеспецифические паттерны экспрессии, ассоциации заболеваний и изоформы miRNA, а также обнаруживать ранее не охарактеризованные miRNA. Доказательства того, что дисрегулируемые miRNA играют роль в таких заболеваниях, как рак, позиционируют miRNA-seq, чтобы потенциально стать важным инструментом в будущем для диагностики и прогнозирования, поскольку затраты продолжают снижаться. Как и другие технологии профилирования miRNA, miRNA-Seq имеет как преимущества (независимость от последовательности, охват), так и недостатки (высокая стоимость, требования к инфраструктуре, длина цикла и потенциальные артефакты ).
МикроРНК (миРНК) представляют собой семейство малых рибонуклеиновых кислот длиной 21-25 нуклеотидов, которые модулируют экспрессию белка посредством деградации транскрипта, ингибирования трансляции , или секвестрирующие транскрипты. Первая миРНК, которая быть обнаруженным, lin-4, был обнаружен в результате генетического мутагенеза для выявления молекулярных элементов, контролирующих постэмбриональное развитие нематоды Caenorhabditis elegans. Ген lin-4 кодировал 22 нуклеотидную РНК с консервативными комплементарными сайтами связывания в 3’-нетранслируемой области транскрипта мРНК и подавлял экспрессию белка LIN-14. В настоящее время считается, что miRNA участвуют в регуляции многих процессов развития и биологических процессов, включая гематопоэз (в Mus musculus ), липидный обмен (в Drosophila melanogaster ) и развитие нейронов (в Caenorhabditis elegans ). Эти открытия потребовали разработки методов, способных идентифицировать и характеризовать miRNAs, такие как miRNA-seq.
Секвенирование микроРНК (miRNA-seq) было разработано, чтобы воспользоваться преимуществами секвенирования следующего поколения или массового параллельного высокопроизводительного секвенирования чтобы найти новые miRNA и профили их экспрессии в данном образце. Секвенирование miRNA само по себе не является новой идеей, в начальных методах секвенирования использовались методы секвенирования по Сэнгеру. Подготовка к секвенированию включала создание библиотек путем клонирования ДНК, обратно транскрибированной с эндогенных малых РНК размером 21-25 п.н., выбранных с помощью колонки и гель-электрофореза. Однако этот метод является исчерпывающим с точки зрения времени и ресурсов, поскольку каждый клон должен быть индивидуально амплифицирован и подготовлен для секвенирования. Этот метод также непреднамеренно отдает предпочтение miRNA с высокой экспрессией. Секвенирование следующего поколения устраняет необходимость в зондах гибридизации, специфичных для последовательности, необходимых для анализа ДНК-микрочипов, а также в трудоемких методах клонирования, требуемых в методе секвенирования по Сэнгеру. Кроме того, платформы секвенирования следующего поколения в методе miRNA-SEQ облегчают секвенирование больших пулов малых РНК за один цикл секвенирования.
miRNA-seq может выполняться с использованием различных платформ секвенирования. Первый анализ малых РНК с использованием методов miRNA-seq исследовал приблизительно 1,4 миллиона малых РНК из модельного растения Arabidopsis thaliana с использованием платформы для секвенирования. Это исследование продемонстрировало потенциал новых технологий высокопроизводительного секвенирования для изучения малых РНК и показало, что геномы генерируют большое количество малых РНК, причем растения являются особенно богатыми источниками малых РНК. В более поздних исследованиях использовались другие технологии секвенирования, такие как исследование C. elegans, которые идентифицировали 18 новых генов miRNA, а также новый класс малых РНК нематод, названный 21U-РНК. В другом исследовании, сравнивающем профили малых РНК опухолей шейки матки человека и нормальной ткани, использовался анализатор генома Illumina (компания) для идентификации 64 новых генов miРНК человека, а также 67 дифференциально экспрессируемых miRNA. Applied Biosystems Платформа для секвенирования SOLiD также использовалась для изучения прогностической ценности miRNAs в обнаружении рака груди человека.
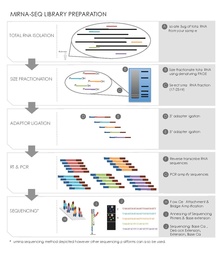 Подготовка библиотеки miRNA
Подготовка библиотеки miRNA Создание библиотеки последовательностей может может выполняться с использованием различных наборов в зависимости от используемой платформы высокопроизводительного секвенирования. Однако существует несколько общих этапов подготовки секвенирования малых РНК.
Выделение полной РНК
В данном образце вся РНК извлекается и выделяется с использованием метода изотиоцианат / фенол / хлороформ (GITC / фенол) или коммерческого продукта. такой как реагент Trizol (Invitrogen ). Начальное количество 50-100 мкг общей РНК, 1 г ткани обычно дает 1 мг общей РНК, обычно требуется для очистки геля и выбора размера. Также измеряется контроль качества РНК, например, запуск чипа РНК на Caliper LabChipGX (Caliper Life Sciences ).
Фракционирование малых РНК по размеру с помощью гель-электрофореза.
Выделенную РНК прогоняли на денатурирующем полиакриламидном геле. Метод визуализации, такой как радиоактивные 5’-32P-меченные олигонуклеотиды вместе с размерной лестницей, используется для идентификации участка геля, содержащего РНК, подходящего размера, уменьшая количество материала, в конечном итоге секвенируемого. Этот этап не обязательно должен выполняться перед этапами лигирования и обратной транскрипции, описанными ниже.
Лигирование
На этапе лигирования к обоим концам малых РНК добавляются адаптеры ДНК, которые действуют как сайты связывания праймеров во время обратной транскрипции и ПЦР амплификация. Аденилированный 3’-адаптер одноцепочечной ДНК, за которым следует 5’-адаптер, лигируют с малыми РНК с помощью лигирующего фермента, такого как РНК-лигаза Т4. Адаптеры также предназначены для захвата малых РНК с 5'-фосфатной группой, характерных микроРНК, а не продуктов деградации РНК с 5'-гидроксильной группой.
Обратная транскрипция и ПЦР-амплификация
На этом этапе преобразуются РНК, лигированные небольшим адаптером в клоны кДНК, используемые в реакции секвенирования. Доступно много коммерческих наборов, которые будут выполнять этот этап с использованием некоторой формы обратной транскриптазы. Затем проводят ПЦР для амплификации пула последовательностей кДНК. Праймеры, разработанные с уникальными нуклеотидными тегами, также можно использовать на этом этапе для создания идентификационных тегов при мультиплексном секвенировании объединенной библиотеки.
Фактическое секвенирование РНК значительно варьируется в зависимости от используемой платформы. Три распространенных платформы секвенирования следующего поколения: пиросеквенирование на платформе 454 Life Sciences, последовательность-синтез на основе полимеразы на платформе Illumina (компания), или секвенирование лигированием на платформе ABI Solid Sequencing.
Центральным в анализе данных miRNA-seq является возможность 1) получать уровни изобилия miRNA из считывания последовательностей, 2) обнаруживать новые miRNA и затем иметь возможность 3) определять дифференциально экспрессируемые miRNA и 4) связанные с ними гены-мишени mRNA.
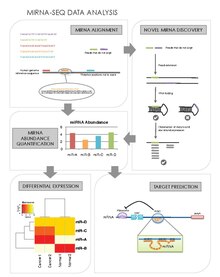 Анализ данных miRNA-seq
Анализ данных miRNA-seq miRNA могут предпочтительно экспрессироваться в определенных типах клеток, тканях, стадиях развития или, в частности, болезненных состояниях, таких как рак. Поскольку глубокое секвенирование (miRNA-seq) генерирует миллионы считываний из данного образца, оно позволяет нам профилировать miRNA; может ли это быть путем количественной оценки их абсолютной численности, чтобы обнаружить их варианты (известные как изомиры ). Обратите внимание, что, учитывая, что средняя длина считывания последовательности больше, чем средняя miRNA (17-25 нуклеотидов), 3 'и 5' концы miRNA должны быть найдены в одном считывании. Существует несколько алгоритмов количественной оценки количества миРНК. Их общие шаги следующие:
Еще одним преимуществом miRNA-seq является то, что он позволяет открывать новые miRNA. РНК, которые могли ускользнуть от традиционных методов скрининга и профилирования. Существует несколько новых алгоритмов обнаружения miRNA. Их общие шаги следующие:
После количественного определения количества miRNA для каждого образца их уровни экспрессии можно сравнивать между образцами. Тогда можно будет идентифицировать miRNA, которые предпочтительно экспрессируются в определенные моменты времени или в определенных тканях или болезненных состояниях. После нормализации количества отображенных считываний между образцами можно использовать множество статистических тестов (например, те, что используются в профилировании экспрессии генов ) для определения дифференциальной экспрессии
Идентификация мишеней мРНК miRNA обеспечит понимание генов или сетей генов, экспрессию которых они регулируют. Общедоступные базы данных предоставляют прогнозы мишеней miRNA. Но чтобы лучше отличать истинно положительные предсказания от ложноположительных, данные miRNA-seq могут быть интегрированы с данными mRNA-seq для наблюдения за функциональными парами miRNA: mRNA. RNA22, TargetScan, miRanda и PicTar - это программное обеспечение, разработанное для этой цели. Список программного обеспечения для прогнозирования приведен здесь. Общие шаги:
Многие миРНК функционируют, чтобы направлять расщепление своих мРНК-мишеней; это особенно верно для растений, и поэтому были разработаны методы высокопроизводительного секвенирования, чтобы воспользоваться этим свойством miRNA путем секвенирования 3'-концов расщепленных или деградированных мРНК. Эти методы известны как секвенирование деградома или PARE. Проверка расщепления-мишени в конкретных мРНК обычно выполняется с использованием модифицированной версии 5 'быстрой амплификации концов кДНК с ген-специфическим праймером.
miRNA-seq выявила новые miRNA, которые ранее ускользнули с помощью традиционных методов профилирования miRNA. Примерами таких результатов являются эмбриональные стволовые клетки, куриные эмбрионы, острый лимфобластный лейкоз, диффузная крупноклеточная В-клеточная лимфома и В-клетки, острый миелоидный лейкоз и рак легких.
Микро РНК являются важными регуляторами почти всех клеточных процессов, таких как выживаемость, пролиферация и дифференцировка. Следовательно, неудивительно, что miRNA участвуют в различных аспектах рака посредством регуляции экспрессии онко- и гена-супрессора опухоли. В сочетании с разработкой высокопроизводительных методов профилирования miRNA были идентифицированы как биомаркеры для классификации рака, ответа на терапию и прогноза. Кроме того, поскольку miRNA регулируют экспрессию генов, они также могут выявлять нарушения в важных регуляторных сетях, которые могут вызывать конкретное заболевание. Ниже приведены несколько применений miRNA в качестве биомаркеров и предикторов заболевания.
| Тип рака | миРНК | Ref. |
|---|---|---|
| Грудь | ||
| Статус ER | miR-26a / b, семейство miR-30, miR-29b, miR-155, miR-342, miR-206, miR-191 | |
| Статус PR | let-7c, miR-29b, miR-26a, семейство miR-30, miR-520g | |
| статус HER2 / neu | miR-520d, miR-181c, miR-302c, miR-376b, miR-30e | |
| Легкое | ||
| Плоскоклеточный и неплоскоклеточный | miR-205 | |
| Мелкоклеточный vs немелкоклеточный | miR-17- 5p, miR-22, miR-24, miR-31 | |
| Желудочный | ||
| Диффузный против кишечного | miR-29b / c, семейство miR-30, miR-135a / b | |
| Эндометриальный | ||
| Эндометриоид против сосочков матки | miR-19a / b, miR-30e-5p, miR-101, miR-452, miR-382, miR-15a, miR-29c | |
| Почечный | ||
| Светлые клетки против папиллярных | miR-424, miR-203, miR-31, miR-126 | |
| Онкоцитома против хромофобных | miR-200c, miR-139-5p | |
| Миелома | ||
| с t (14; 16) | miR-1, miR-133a | |
| с t (4; 14) | miR-203, miR-155, miR-375 | |
| с t (11; 14) | miR-125a, miR-650, miR-184 | |
| Острый миелоидный лейкоз | ||
| с t (15; 17) | miR -382, miR-134, miR-376a, miR-127, mi R-299-5p, miR-323 | |
| с t (8; 21) или inv (16) | let-7b / c, miR-127 | |
| с мутациями NPM1 | miR-10a / b, let-7, miR-29, miR-204, miR-128a, miR-196a / b | |
| с ITD FLT3 | miR-155 | |
| Хронический лимфолейкоз | ||
| Уровни ZAP-70 и статус IgVH | miR-15a, miR-195, miR-221, miR-155, miR-23b | |
| Меланома | ||
| с BRAF V600E | miR-193a, miR-338, miR-565 | |
| лимфома | ||
| диффузная крупноклеточная B-лимфома | has-miR-128, has-miR-129-3p, has-miR-152, имеет -miR-155, has-miR-185, has-miR-193a-5p, has-miR-196b, has-miR-199b-3p, has-miR-20b, has-miR-23a, has-miR-27a, has-miR-28-5p, has-miR-301a, has-miR-331-3p, has-miR-365, has-miR-625, has-miR-9 | |
Это неполный список miRNAs, вовлеченные в эти злокачественные новообразования.
Недостатки использования miRNA-seq по сравнению с другими методами профилирования miRNA состоят в том, что он более дорогой, обычно требует большего количества общей РНК, требует значительного амплификация и требует больше времени, чем методы микроматрицы и КПЦР. Кроме того, методы подготовки библиотеки miRNA-seq, по-видимому, систематически предпочтительно представляют комплемент miRNA, и это препятствует точному определению количества miRNA. В то же время этот подход не зависит от гибридизации и, следовательно, не требует априорной информации о последовательности. Благодаря этому можно получить последовательности новых miRNA и изоформ miRNA (isoMirs), различать последовательно похожие miRNA и идентифицировать точечные мутации.
| qPCR | Микроматрица | Секвенирование | |
|---|---|---|---|
| Время обработки | ~ 6 часов | ~ 2 дня | 1-2 недель |
| Общая требуемая РНК | 500 нг | 100-1000 нг | 500-5000 нг |
| Обнаружен динамический диапазон | Шесть порядков величина | Четыре порядка величины | Пять или более порядков величины |
| Инфраструктура и технические требования | Немного | Умеренная | Существенная |
| Стоимость образца (долл. США) | 400 долл. США | 250–350 долл. США | 500–700 долл. США |