
Войти
| Синдром Пфайффера | |
|---|---|
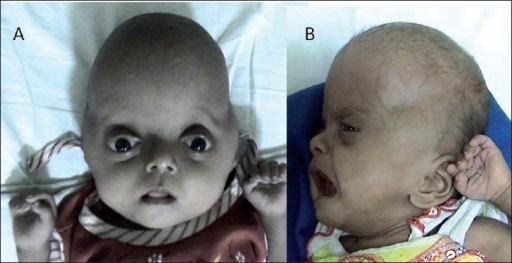 | |
| Синдром Пфайффера типа 2 с черепом в форме клеверного листа и двусторонним проптозом до и после операции | |
| Специальность | Ревматология |
| Причины | Генетические |
| Частота | 1 на 100 000 рождений |
Синдром Пфайффера встречается редко генетическое заболевание, характеризующееся преждевременным сращением некоторых костей черепа (краниосиностоз ), которое влияет на форму головы и лица. Кроме того, синдром включает аномалии рук (например, широкие и искривленные большие пальцы рук) и ступни (например, широкие и искривленные большие пальцы ног).
Синдром Пфайффера вызывается мутациями в рецепторах фактора роста фибробластов FGFR1 и FGFR2. Синдром подразделяется на три типа: тип 1 (классический синдром Пфайффера) более мягкий и вызван мутациями в любом из генов, а типы 2 и 3 более тяжелые, часто приводящие к смерти в младенчестве, вызванные мутациями в FGFR2.
От синдрома нет лекарства. Лечение является поддерживающим и часто включает в себя хирургическое вмешательство в первые годы жизни для исправления деформации черепа и респираторной функции. Большинство людей с синдромом Пфайффера типа 1 имеют нормальный интеллект и продолжительность жизни, тогда как типы 2 и 3 обычно приводят к нарушениям развития нервной системы и ранней смерти.
Синдром Пфайффера встречается примерно у 1 из 100 000 новорожденных. Синдром назван в честь немецкого генетика Рудольфа Артура Пфайффера (1931–2012), описавшего его в 1964 году.
Многие из характерные черты лица являются результатом преждевременного сращения костей черепа (краниосиностоз ). Голова не может нормально расти, что приводит к высокому выступающему лбу (туррибрахицефалия ) и глазам, которые кажутся выпуклыми (проптоз ) и широко расставленными (гипертелоризмом. ). Кроме того, имеется недоразвитая верхняя челюсть (гипоплазия верхней челюсти ). Около 50 процентов детей с синдромом Пфайффера страдают потерей слуха; Также часто встречаются стоматологические проблемы.
У людей с синдромом Пфайффера большие пальцы рук и пальцы на ногах широкие и согнуты в сторону от других пальцев (варусный Pollex и варусный палец ). Также часто встречаются необычно короткие пальцы рук и ног (брахидактилия ), и между пальцами может быть перепонка или слияние (синдактилия ).
синдром Пфайффера тесно связан с мутации рецептора фактора роста фибробластов 1 (FGFR1) на хромосоме 8 или гена рецептора фактора роста фибробластов 2 (FGFR2) на хромосоме 10. Эти гены кодируют рецепторы фактора роста фибробластов, которые важны для нормального развития костей. Возраст отца считается фактором риска для спорадических случаев синдрома Пфайффера, вызванного увеличение количества мутаций в сперматозоидах по мере старения мужчин.
Наиболее широко принятая клиническая классификация синдрома Пфайффера была опубликована М. Майклом Коэном в 1993 году. Коэн разделил синдром на три, возможно, частично совпадающих типа, каждый из которых характеризуется широкими большими пальцами рук, широкими большими пальцами ног, брахидактилией и позой. вероятно синдактилия :
Ключевой проблемой является раннее слияние череп, который можно исправить с помощью серии хирургических процедур, часто в течение первых трех месяцев после рождения. Для исправления респираторных и лицевых деформаций необходимы более поздние операции.
Дети с синдромом Пфайффера 2 и 3 типа "имеют более высокий риск нарушений психического развития и сокращают продолжительность жизни продолжительность беременности », чем у детей с синдромом Пфайффера типа 1, но при лечении возможны благоприятные исходы. В тяжелых случаях респираторные и неврологические осложнения часто приводят к преждевременной смерти.
Синдром назван в честь немецкого генетика Рудольфа Артура Пфайффера (1931–2012). В 1964 году Пфайффер описал восемь человек в трех поколениях семьи, у которых были аномалии головы, рук и ног (акроцефалосиндактилия ), которые были унаследованы по аутосомно-доминантному типу.
| journal =()| Классификация | D |
|---|---|
| Внешние ресурсы |