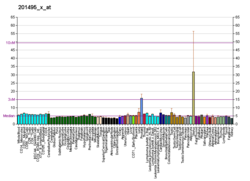
| MYH11 |
|---|
 |
| Идентификаторы |
|---|
| Псевдонимы | MYH11, AAT4, FAA4, SMHC, SMMHC, миозин, тяжелая цепь 11, гладкие мышцы, тяжелая цепь миозина 11, VSCM2 |
| Внешние идентификаторы | OMIM : 160745 MGI : 102643 HomoloGene : 128512 GeneCards : MYH11 |
|
| Расположение гена ( Мышь ) |
|---|
 | | Chr. | Хромосома 16 (мышь) |    | | Группа | 16 A1 | 16 9,71 см | Начинать | 14 194 535 п.н. | | Конец | 14 291 372 п.н. | |
|
|
| Ортологи |
|---|
| Разновидность | Человек | Мышь |
| Entrez | | |
| Ансамбль | | |
| UniProt | | |
| RefSeq (мРНК) | | |
| RefSeq (белок) | | |
| Расположение (UCSC) | Chr 16: 15,7 - 15,86 Мб | Chr 16: 14.19 - 14.29 Мб |
| PubMed поиск | | |
| Викиданные |
|
Миозин-11 представляет собой белок, который у человека кодируется MYH11 гена.
СОДЕРЖАНИЕ
- 1 Функция
- 2 Клиническое значение
- 3 Острый миелоидный лейкоз
- 4 Рак кишечника
- 5 ссылки
- 6 Внешние ссылки
- 7 Дальнейшее чтение
Функция
Миозин-11 - это миозин гладких мышц, принадлежащий к семейству тяжелых цепей миозина. Миозин-11 представляет собой субъединицу гексамерного белка, которая состоит из двух субъединиц тяжелой цепи и двух пар неидентичных субъединиц легкой цепи.
Это основной сократительный белок, преобразующий химическую энергию в механическую посредством гидролиза АТФ.
Альтернативный сплайсинг генерирует изоформы, которые дифференциально экспрессируются, причем соотношение меняется во время созревания мышечных клеток.
Клиническое значение
Аневризмы грудной аорты, приводящие к острому расслоению аорты (TAAD), могут быть унаследованы изолированно или в сочетании с генетическими синдромами, такими как синдром Марфана и синдром Лойса-Дитца. Когда TAAD возникает при отсутствии синдромных признаков, он наследуется по аутосомно-доминантному типу с пониженной пенетрантностью и вариабельной экспрессией, заболевание называется семейным TAAD. Семейный TAAD демонстрирует значительную клиническую и генетическую гетерогенность. Мутации в MYH11 были описаны у людей с TAAD с открытым артериальным протоком (PDA). Примерно 4% лиц с TAAD имеют мутации в TGFBR2 и примерно 1-2% имеют мутации в TGFBR1 или MYH11. Кроме того, мутации FBN1 также были зарегистрированы у лиц с TAAD. Недавно сообщалось о мутациях в гене SMAD3 у пациентов с синдромальной формой аневризмы и расслоения аорты с ранним началом остеоартрита. Считается, что мутации SMAD3 составляют примерно 2% семейного TAAD. Кроме того, считается, что мутации в гене ACTA2 составляют примерно 10–14% семейного TAAD.
Острый миелоидный лейкоз
Ген, кодирующий человеческий ортолог крысиного NUDE1, транскрибируется с обратной цепи этого гена, и его 3'-конец перекрывается с концом последнего. Перицентрическая инверсия хромосомы 16 [inv (16) (p13q22)] дает химерный транскрипт, который кодирует белок, состоящий из первых 165 остатков с N-конца бета-фактора связывания ядра в слиянии с C-концевой частью тяжелая цепь миозина гладких мышц. Эта хромосомная перестройка связана с острым миелоидным лейкозом подтипа M4Eo.
Рак кишечника
Мутации MYH11, по-видимому, способствуют развитию рака кишечника человека.
использованная литература
внешние ссылки
дальнейшее чтение
- Бабу Г.Дж., Уоршоу Д.М., Периасами М. (2000). «Изоформы тяжелых цепей гладкомышечного миозина и их роль в физиологии мышц». Microsc. Res. Tech. 50 (6): 532–40. DOI : 10.1002 / 1097-0029 (20000915) 50: 6 lt;532:: АИД-JEMT10gt; 3.0.CO; 2-Е. PMID 10998642.
- Айкава М., Сивам П.Н., Куро-о М. и др. (1993). "Изоформы тяжелой цепи миозина гладких мышц человека как молекулярные маркеры развития сосудов и атеросклероза". Circ. Res. 73 (6): 1000–12. DOI : 10.1161 / 01.res.73.6.1000. PMID 7916668.
- van der Reijden BA, Dauwerse JG, Wessels JW и др. (1993). «Ген пептида миозина нарушается inv (16) (p13q22) при остром нелимфоцитарном лейкозе M4Eo». Кровь. 82 (10): 2948–52. DOI : 10.1182 / blood.V82.10.2948.2948. PMID 8219185.
- Дэн З., Лю П., Марлтон П. и др. (1994). «Локус тяжелой цепи гладкомышечного миозина (MYH11) отображается на 16p13.13-p13.12 и устанавливает новую область консервативной синтении между 16p человека и 16 мыши». Геномика. 18 (1): 156–9. DOI : 10.1006 / geno.1993.1443. ЛВП : 2027,42 / 30537. PMID 8276405.
- Shoeman RL, Sachse C, Höner B и др. (1993). «Расщепление белков цитоскелета и саркомера человека и мыши протеазой вируса иммунодефицита человека типа 1. Актин, десмин, миозин и тропомиозин». Являюсь. J. Pathol. 142 (1): 221–30. PMC 1886840. PMID 8424456.
- New L, Jiang Y, Zhao M, et al. (1998). «PRAK, новая протеинкиназа, регулируемая киназой p38 MAP». EMBO J. 17 (12): 3372–84. DOI : 10.1093 / emboj / 17.12.3372. PMC 1170675. PMID 9628874.
- Танака Ю., Фуджи М., Хаяши К. и др. (1998). «Химерный белок PEBP2 beta / CBF beta-SMMHC дезорганизует цитоплазматические стрессовые волокна и ингибирует активацию транскрипции». Онкоген. 17 (6): 699–708. DOI : 10.1038 / sj.onc.1201985. PMID 9715271.
- Нагасе Т., Исикава К., Суяма М. и др. (1999). «Прогнозирование кодирующих последовательностей неидентифицированных генов человека. XII. Полные последовательности 100 новых клонов кДНК из мозга, которые кодируют большие белки in vitro». ДНК Res. 5 (6): 355–64. DOI : 10.1093 / dnares / 5.6.355. PMID 10048485.
- Лофтус Б.Дж., Ким Ю.Дж., Снеддон В.П. и др. (1999). «Дупликации генома и другие особенности в 12 Mb последовательности ДНК из хромосомы 16p и 16q человека». Геномика. 60 (3): 295–308. DOI : 10.1006 / geno.1999.5927. PMID 10493829.
- Цучио Ю., Наито С., Ногами А. и др. (2000). «Интракоронарные уровни тяжелых цепей миозина гладких мышц сыворотки после ЧТКА могут предсказать рестеноз». Японский журнал сердца. 41 (2): 131–40. DOI : 10,1536 / jhj.41.131. PMID 10850529.
- Лин В.К., Ван Д., Ли И.Л. и др. (2000). «Экспрессия гена тяжелой цепи миозина в нормальной и гиперпластической ткани предстательной железы человека». Простаты. 44 (3): 193–203. DOI : 10.1002 / 1097-0045 (20000801) 44: 3 lt;193:: АИД-PROS3gt; 3.0.CO; 2-А. PMID 10906735.
- Мелони I, Рубеньи П., Де Алоэ Дж. И др. (2001). «Эластическая псевдоксантома: точечные мутации в гене ABCC6 и большая делеция, включая также ABCC1 и MYH11». Гм. Мутат. 18 (1): 85. DOI : 10.1002 / humu.1157. PMID 11439001. S2CID 23970082.
- Кунду М., Чен А., Андерсон С. и др. (2002). «Роль Cbfb в гемопоэзе и нарушениях, возникающих в результате экспрессии лейкемогенного гена слияния Cbfb-MYH11». Кровь. 100 (7): 2449–56. DOI : 10,1182 / кровь 2002-04-1064. PMID 12239155.
- Strausberg RL, Feingold EA, Grouse LH, et al. (2003). «Создание и первоначальный анализ более 15 000 полноразмерных последовательностей кДНК человека и мыши». Proc. Natl. Акад. Sci. США. 99 (26): 16899–903. DOI : 10.1073 / pnas.242603899. PMC 139241. PMID 12477932.
- Ота Т., Сузуки Ю., Нисикава Т. и др. (2004). «Полное секвенирование и характеристика 21 243 полноразмерных кДНК человека». Nat. Genet. 36 (1): 40–5. DOI : 10,1038 / нг1285. PMID 14702039.
- Ландретт С.Ф., Куо Ю.Х., Хенсен К. и др. (2005). «Plag1 и Plagl2 - онкогены, которые вызывают острый миелоидный лейкоз в сотрудничестве с Cbfb-MYH11». Кровь. 105 (7): 2900–7. DOI : 10.1182 / кровь-2004-09-3630. PMID 15585652.
- Léguillette R, Gil FR, Zitouni N, et al. (2005). «(+) Вставить экспрессию изоформы тяжелой цепи миозина гладких мышц (SM-B) в ткани человека». Являюсь. J. Physiol., Cell Physiol. 289 (5): C1277–85. DOI : 10,1152 / ajpcell.00244.2004. PMID 16000639.
- Чжу Л., Вранкс Р., Хау Ван Киен П. и др. (2006). «Мутации в тяжелой цепи 11 миозина вызывают синдром, связанный с аневризмой грудной аорты / расслоением аорты и открытым артериальным протоком». Nat. Genet. 38 (3): 343–9. DOI : 10.1038 / ng1721. PMID 16444274. S2CID 2890964.