
Войти
 Рабочий процесс секвенирования экзома: часть 1.
Рабочий процесс секвенирования экзома: часть 1. Секвенирование экзома, также известное как секвенирование всего экзома (WES ), представляет собой геномный метод секвенирования всех кодирующих белки областей гены в геноме (известном как экзом ). Он состоит из двух шагов: первый шаг - выбрать только подмножество ДНК, которое кодирует белки. Эти области известны как экзоны - люди имеют около 180000 экзонов, составляющих около 1% генома человека, или около 30 миллионов пар оснований. Второй шаг - секвенирование экзонной ДНК с использованием любой высокопроизводительной технологии секвенирования ДНК.
Цель этого подхода - идентифицировать генетические варианты, которые изменяют последовательности белков, и сделать это на гораздо более низкая стоимость, чем секвенирование всего генома. Поскольку эти варианты могут быть ответственны как за менделевские, так и за распространенные полигенные заболевания, такие как болезнь Альцгеймера, полное секвенирование экзома применялось как в академических исследованиях, так и в качестве клиническая диагностика.
 Рабочий процесс секвенирования экзома: часть 2.
Рабочий процесс секвенирования экзома: часть 2. Секвенирование экзома особенно эффективно при изучении редких менделевских заболеваний, потому что это эффективный способ идентифицировать генетические варианты во всех генах человека. Эти заболевания чаще всего вызываются очень редкими генетическими вариантами, которые присутствуют только у небольшого числа людей; Напротив, такие методы, как массивы SNP, могут обнаруживать только общие генетические варианты, которые являются общими для многих людей в более широкой популяции. Более того, поскольку варианты, вызывающие серьезные заболевания, гораздо более вероятно (но ни в коем случае не исключительно) присутствуют в кодирующей последовательности белка, сосредоточение внимания на этом 1% стоит гораздо меньше, чем секвенирование всего генома, но все же обнаруживает высокую выход соответствующих вариантов.
В прошлом клинические генетические тесты выбирались на основе клинических проявлений пациента (т. Е. Фокусировались на одном гене или небольшом количестве, которое, как известно, связано с определенным синдромом), или изучали только определенные типы вариаций (например, сравнительная геномная гибридизация ), но дала окончательный генетический диагноз менее чем у половины всех пациентов. В настоящее время секвенирование экзома все чаще используется в дополнение к этим другим тестам: как для поиска мутаций в генах, которые, как известно, вызывают заболевание, так и для выявления новых генов путем сравнения экзомов от пациентов со схожими характеристиками.
Методы обогащения мишеней позволяют выборочно захватывать интересующие области генома из образца ДНК до секвенирования. Со времени первоначального описания метода прямого геномного отбора (DGS) в 2005 году было разработано несколько стратегий обогащения мишеней.
Хотя многие методы были описаны для целевого захвата, только некоторые из них были расширены для захвата целые экзомы. Первой стратегией целевого обогащения, которая была применена к секвенированию всего экзома, был метод гибридного захвата на основе массива в 2007 году, но в последние годы захват в растворе приобрел популярность.
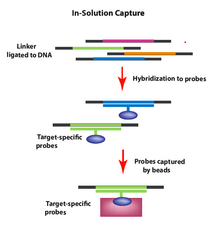 Улавливание в растворе.
Улавливание в растворе. Микроматрицы содержат одноцепочечные олигонуклеотиды с последовательностями из генома человека для мозаичного размещения интересующей области, прикрепленной к поверхности. Геномная ДНК разрезается с образованием двухцепочечных фрагментов. Фрагменты подвергаются репарации на концах с образованием тупых концов, и добавляются адаптеры с универсальными последовательностями прайминга. Эти фрагменты гибридизируются с олигонуклеотидами на микрочипе. Негибридизированные фрагменты смываются и желаемые фрагменты элюируются. Затем фрагменты амплифицируются с помощью ПЦР.
Roche NimbleGen была первой, кто взял оригинальную технологию DGS и адаптировал ее для секвенирования следующего поколения. Они разработали массив 2.1M Sequence Capture Human Exome для захвата ~ 180 000 кодирующих экзонов. Этот метод экономит время и экономичен по сравнению с методами на основе ПЦР. Массив захвата Agilent и массив сравнительной геномной гибридизации - это другие методы, которые можно использовать для гибридного захвата целевых последовательностей. Ограничения этого метода включают необходимость в дорогостоящем оборудовании, а также в относительно большом количестве ДНК.
Для захвата интересующих участков генома с помощью захвата в растворе пул стандартных олигонуклеотидов (зондов) синтезируют и гибридизируют в растворе с образцом фрагментированной геномной ДНК. Зонды (помеченные шариками) селективно гибридизуются с интересующими областями генома, после чего шарики (теперь включающие интересующие фрагменты ДНК) могут быть сняты и промыты для удаления излишков материала. Затем гранулы удаляются, и геномные фрагменты могут быть секвенированы, что позволяет селективно секвенировать ДНК интересующих геномных областей (например, экзонов).
Этот метод был разработан для улучшения метода гибридизационного захвата-обогащения. При захвате раствора (в отличие от гибридного захвата) существует избыток зондов для целевых областей, представляющих интерес, по сравнению с количеством требуемой матрицы. Оптимальный размер мишени составляет около 3,5 мегабайт и обеспечивает превосходный охват последовательностей целевых областей. Предпочтительный метод зависит от нескольких факторов, включая: количество пар оснований в интересующей области, требования к считыванию на мишени, оборудование в доме и т. Д.
доступно множество платформ секвенирования следующего поколения секвенирования, появившихся после классических методологий секвенирования Сэнгера. Другие платформы включают секвенсор Roche 454 и Life Technologies системы SOLiD, Life Technologies Ion Torrent и Illumina's Illumina Genome Analyzer II (несуществующий) и последующие инструменты серии Illumina MiSeq, HiSeq и NovaSeq, все из которых могут использоваться для массового параллельного секвенирования экзома. Эти системы NGS с «коротким считыванием» особенно хорошо подходят для анализа многих относительно коротких участков последовательности ДНК, обнаруженных в экзонах человека.
Есть несколько доступных технологий, которые идентифицируют генетические варианты. Каждая технология имеет преимущества и недостатки с точки зрения технических и финансовых факторов. Двумя такими технологиями являются микроматрицы и полногеномное секвенирование.
Микроматрицы используют зонды гибридизации для проверки распространенности известных последовательностей ДНК, поэтому их нельзя использовать для выявления неожиданных генетических изменений. Напротив, технологии высокопроизводительного секвенирования, используемые при секвенировании экзома, непосредственно обеспечивают нуклеотидные последовательности ДНК в тысячах тестируемых экзонных локусов. Следовательно, WES устраняет некоторые из существующих ограничений гибридизации генотипирования массивов.
Хотя секвенирование экзома дороже технологий на основе гибридизации для каждого образца, его стоимость снижается из-за снижения стоимости и увеличения производительности секвенирования всего генома.
Секвенирование экзома позволяет идентифицировать только те варианты, обнаруженные в кодирующей области генов, которые влияют на функцию белка. Он не может идентифицировать структурные и некодирующие варианты, связанные с заболеванием, которые можно найти с помощью других методов, таких как полногеномное секвенирование. Остается 99% генома человека, не охваченного секвенированием экзома. В настоящее время секвенирование всего генома редко применяется в клинических условиях из-за высоких затрат и времени, связанных с секвенированием полных геномов. Секвенирование экзома позволяет секвенировать части генома, по крайней мере, в 20 раз большем количестве образцов по сравнению с секвенированием всего генома при тех же затратах. Для перевода идентифицированных редких вариантов в клинику размер выборки и способность интерпретировать результаты для постановки клинического диагноза указывают на то, что с учетом современных знаний в области генетики секвенирование экзома может быть наиболее ценным.
Статистический анализ большого количества данных, полученных с помощью подходов к секвенированию, является сложной задачей. Даже при секвенировании только экзомов людей генерируется большое количество данных и информации о последовательностях, что требует значительного анализа данных. Проблемы, связанные с анализом этих данных, включают изменения в программах, используемых для выравнивания и сборки считываний последовательностей. Различные технологии секвенирования также имеют разную частоту ошибок и генерируют разную длину чтения, что может создавать проблемы при сравнении результатов с разных платформ секвенирования.
Ложноположительные и ложноотрицательные результаты связаны с подходами к геномному пересеквенированию и являются критическими проблемами. Было разработано несколько стратегий для улучшения качества данных экзома, таких как:
Редкие рецессивные заболевания не имели бы однонуклеотидных полиморфизмов (SNP) в общедоступных базах данных, таких как dbSNP. Более распространенные рецессивные фенотипы могут иметь вызывающие заболевание варианты, описанные в dbSNP. Например, наиболее распространенный вариант муковисцидоза имеет частоту аллелей около 3% в большинстве популяций. Скрининг таких вариантов может ошибочно исключить такие гены из рассмотрения. Гены рецессивных расстройств обычно легче идентифицировать, чем доминантные расстройства, потому что у этих генов меньше шансов иметь более одного редкого несинонимичного варианта. Система, которая проверяет общие генетические варианты, полагается на dbSNP, который может не иметь точной информации об изменении аллелей. Использование списков общих вариаций от исследуемого экзома или индивидуума, секвенированного по всему геному, было бы более надежным. Проблема в этом подходе состоит в том, что по мере увеличения числа секвенированных экзомов dbSNP также будет увеличиваться в количестве необычных вариантов. Потребуется разработать пороговые значения для определения общих вариантов, которые вряд ли будут связаны с фенотипом заболевания.
Генетическая гетерогенность и популяция этническая принадлежность также являются серьезными ограничениями, поскольку они могут увеличивать число ложноположительных и ложноотрицательных результатов, которые затруднят идентификацию генов-кандидатов. Конечно, можно снизить строгость пороговых значений при наличии неоднородности и этнической принадлежности, однако это также снизит возможность обнаружения вариантов. Использование подхода «сначала генотип» для идентификации генов-кандидатов также может предложить решение для преодоления этих ограничений.
Новые технологии в геномике изменили подход исследователей как к фундаментальным, так и к трансляционным исследованиям. С помощью таких подходов, как секвенирование экзома, можно значительно улучшить данные, полученные из отдельных геномов, что поставило ряд вопросов о том, как обращаться с огромным объемом информации. Следует ли предоставить участникам этих исследований доступ к информации о последовательности? Следует ли передавать эту информацию страховым компаниям? Эти данные могут привести к неожиданным результатам и усложнить клиническое применение и пользу для пациентов. Эта область геномики по-прежнему остается сложной задачей, и исследователи ищут способы решения этих вопросов.
Используя секвенирование экзома, исследования с фиксированной стоимостью могут упорядочить образцы до гораздо более высокой глубины, чем можно было бы достичь с помощью секвенирования всего генома. Эта дополнительная глубина делает секвенирование экзома хорошо подходящим для нескольких приложений, которым требуются надежные вызовы вариантов.
Текущие исследования ассоциации сосредоточены на общих вариациях в геноме, поскольку их легче всего идентифицировать с помощью наших текущих анализов. Однако в исследованиях генов-кандидатов было обнаружено, что вызывающие заболевание варианты с большим эффектом лежат в пределах экзом, и из-за отрицательного отбора обнаруживаются с гораздо более низкими частотами аллелей и могут оставаться нетипизированными в текущих стандартных анализах генотипирования. Полногеномное секвенирование - это потенциальный метод анализа нового варианта генома. Однако при сложных расстройствах (таких как аутизм) считается, что большое количество генов связано с риском заболевания. Эта неоднородность основного риска означает, что для открытия генов требуются очень большие размеры выборки, и, следовательно, секвенирование всего генома не является особенно рентабельным. Эта проблема размера выборки решается разработкой новых передовых аналитических методов, которые эффективно картируют гены болезней, несмотря на то, что генетические мутации редки на уровне вариантов. Кроме того, варианты в кодирующих областях были гораздо более тщательно изучены, и их функциональное значение гораздо проще получить, что делает практическое применение вариантов в целевой области экзома более доступным.
Секвенирование экзома в редком варианте Открытие гена остается очень активной и постоянной областью исследований: на сегодняшний день обнаружено несколько связанных генов, но появляется все больше свидетельств того, что это значительная нагрузка риска наблюдается по наборам генов.
При менделевских расстройствах с большим эффектом полученные данные предполагают, что в основе всего состояния лежит один или очень небольшое количество вариантов в кодирующих генах. Из-за серьезности этих нарушений предполагается, что несколько причинных вариантов являются чрезвычайно редкими или новыми в популяции и могут быть пропущены любым стандартным анализом генотипирования. Секвенирование экзома обеспечивает вызовы вариантов с высоким охватом в кодирующих областях, которые необходимы для отделения истинных вариантов от шума. Успешная модель открытия менделевских генов включает открытие вариантов de novo с использованием трио-секвенирования, при котором родители и пробанд генотипированы.
В исследовании, опубликованном в сентябре 2009 года, обсуждался эксперимент, подтверждающий концепцию, чтобы определить, можно ли идентифицировать причинные генетические варианты с помощью секвенирования экзома. Они секвенировали четырех человек с синдромом Фримена-Шелдона (FSS) (OMIM 193700), редким аутосомно-доминантным заболеванием, которое, как известно, вызвано мутацией в гене MYH3. Восемь HapMap индивидуумов также были секвенированы для удаления общих вариантов с целью определения причинного гена для FSS. После исключения распространенных вариантов авторы смогли идентифицировать MYH3, что подтверждает, что секвенирование экзома может использоваться для выявления причинных вариантов редких заболеваний. Это было первое опубликованное исследование, в котором секвенирование экзома использовалось как подход к идентификации неизвестного причинного гена для редкого менделевского расстройства.
Впоследствии другая группа сообщила об успешном клиническом диагнозе пациента с подозрением на синдром Барттера турецкого происхождения. Синдром Барттера - это болезнь почечной недостаточности соли. Секвенирование экзома выявило неожиданную хорошо консервативную рецессивную мутацию в гене под названием SLC26A3, который связан с врожденной хлоридной диареей (CLD). Этот молекулярный диагноз ХЗЛ был подтвержден лечащим врачом. Этот пример предоставил доказательство концепции использования секвенирования всего экзома в качестве клинического инструмента при оценке пациентов с недиагностированными генетическими заболеваниями. Этот отчет считается первым применением технологии секвенирования нового поколения для молекулярной диагностики пациента.
Второй отчет был проведен по секвенированию экзома людей с менделевским расстройством, известным как синдром Миллера (MIM # 263750), редким заболеванием аутосомно-рецессивным наследованием. Были изучены два брата и сестры и два неродственных человека с синдромом Миллера. Они рассмотрели варианты, которые потенциально могут быть патогенными, такие как несинонимичные мутации, акцепторные и донорские сайты сплайсинга, а также короткие кодирующие вставки или делеции. Поскольку синдром Миллера - редкое заболевание, предполагается, что причинный вариант ранее не был идентифицирован. Предыдущие исследования секвенирования экзома распространенных однонуклеотидных полиморфизмов (SNP) в общедоступных базах данных SNP были использованы для дальнейшего исключения генов-кандидатов. После исключения этих генов авторы обнаружили мутации в DHODH, которые были характерны для людей с синдромом Миллера. Каждый человек с синдромом Миллера представлял собой соединение гетерозигот для мутаций DHODH, которые были унаследованы, поскольку было обнаружено, что каждый родитель пораженного человека является носителем.
Это было впервые было показано, что секвенирование экзома позволяет идентифицировать новый ген, ответственный за редкую менделевскую болезнь. Это захватывающее открытие демонстрирует, что секвенирование экзома может определять местонахождение генов, вызывающих сложные заболевания, что ранее было невозможно из-за ограничений традиционных методов. Целевой захват и массовое параллельное секвенирование представляют собой экономичную, воспроизводимую и надежную стратегию с высокой чувствительностью и специфичностью для обнаружения вариантов, вызывающих изменения кодирования белков в отдельных геномах человека.
Секвенирование экзома может использоваться для диагностики генетической причины заболевания у пациента. Выявление мутации (мутаций) в гене основного заболевания может иметь большое значение для диагностических и терапевтических подходов, может служить ориентиром для прогнозирования естественного течения болезни и дает возможность тестировать членов семьи из группы риска. Существует множество факторов, которые делают секвенирование экзома лучше анализа отдельного гена, включая способность идентифицировать мутации в генах, которые не были протестированы из-за атипичной клинической картины, или способность идентифицировать клинические случаи, когда мутации из разных генов способствуют различным фенотипам в тот же пациент.
После диагностики генетической причины заболевания эта информация может помочь в выборе подходящего лечения. Впервые эта стратегия была успешно реализована в клинике при лечении младенца с воспалительным заболеванием кишечника. Ранее использовался ряд традиционных диагностических методов, но результаты не могли объяснить симптомы младенца. Анализ данных секвенирования экзома выявил мутацию в гене XIAP. Знание функции этого гена послужило основой для лечения младенца, что привело к трансплантации костного мозга, которая вылечила ребенка от болезни.
Исследователи использовали секвенирование экзома, чтобы определить основную мутацию у пациента с синдромом Барттера и врожденной хлоридной диареей. Группа Билгулара также использовала секвенирование экзома и определила основную мутацию у пациента с тяжелыми пороками развития мозга, заявив: «[Эти результаты] подчеркивают использование полного секвенирования экзома для определения локусов болезни в условиях, в которых традиционные методы оказались сложными... Наши результаты продемонстрировать, что эта технология будет особенно ценной для открытия генов в тех условиях, в которых картирование было затруднено из-за гетерогенности локусов и неопределенности в отношении границ диагностической классификации, указывая на светлое будущее для ее широкого применения в медицине ".
Исследователи из Университета Кейптауна, Южная Африка, использовали секвенирование экзома, чтобы обнаружить генетическую мутацию CDH2 как основную причину генетического нарушения, известного как аритмогенная кардиомиопатия правого желудочка (ARVC), которая увеличивает риск сердечных заболеваний и остановки сердца. [1]
Несколько компаний предложили потребителям секвенирование экзома.
Knome была первой компанией, предложившей потребителям услуги по секвенированию экзома стоимостью в несколько тысяч долларов. Позже 23andMe запустил пилотную программу WES, о которой было объявлено в сентябре 2011 года и которая была прекращена в 2012 году. Потребители могли получить данные экзома за 999 долларов. Компания предоставила необработанные данные и не предложила анализ.
В ноябре 2012 года DNADTC, подразделение Gene by Gene, начало предлагать экзомы с 80-кратным покрытием и начальной ценой 695 долларов. Эта цена за веб-сайт DNADTC в настоящее время составляет 895 долларов США. В октябре 2013 года BGI объявила о поощрении персонального секвенирования всего экзома с 50-кратным покрытием за 499 долларов. В июне 2016 года Genos смогла достичь еще более низкой цены в 399 долларов за сертифицированный CLIA потребительский экзом 75X, секвенированный из слюны.