
Войти
| Спинальные мышечные атрофии | |
|---|---|
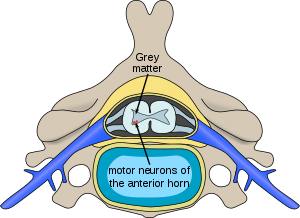 | |
| Расположение нейронов, пораженных спинальными мышечными атрофиями | |
| Специальность | Неврология |
Спинальные мышечные атрофии (SMA ) - генетически и клинически гетерогенная группа редких изнурительных заболеваний, характеризующихся дегенерацией нижние двигательные нейроны (нейронные клетки, расположенные в переднем роге спинного мозга ) и последующая атрофия (истощение) различных группы мышц тела. В то время как некоторые СМА приводят к ранней младенческой смерти, другие болезни этой группы позволяют вести нормальную взрослую жизнь только с легкой слабостью.
В зависимости от типа пораженных мышц, спинальные мышечные атрофии можно разделить на:
Принимая во внимание распространенность, спинальные мышечные атрофии традиционно делятся на:
Более подробная классификация основана на гене, связанном с заболеванием (если он идентифицирован), и представлена в таблице ниже.
| Группа | Имя. Альтернативные названия | OMIM | Ген | Локус | Характеристики | |
|---|---|---|---|---|---|---|
| SMA | Спинальная мышечная атрофия (SMA)
| 253300. 253550. 253400. 271150 | SMN1 | 5q13.2 | Аутосомно-рецессивный | Поражает в первую очередь проксимальные мышцы у людей всех возрастов, прогрессирует, относительно часто |
| XLSMA | Х-сцепленная спинномозговая атрофия 1 типа (SMAX1)
| 313200 | NR3C4 | Xq12 | Х-сцепленный рецессивный | Поражает в первую очередь бульбарные мышцы, а также сенсорные нервы в основном у взрослых мужчин, прогрессирующая |
Х-сцепленная спинальная мышечная атрофия 2 типа (SMAX2)
| 301830 | UBA1 | Xp11.23 | Х-сцепленный рецессивный | Характеризуется переломами костей, поражает в основном дистальные мышцы новорожденного мальчики, обычно со смертельным исходом в младенчестве | |
(SMAX3)
| 300489 | ATP7A | Xq21.1 | X-сцепленная рецессивная | Поражает дистальные мышцы всех конечностей в основном у мальчиков, медленно прогрессирующая | |
| DSMA | Дистальная мышечная атрофия позвоночника 1 типа (DSMA1)
| 604320 | IGHMBP2 | 11q13.3 | Аутосомно-рецессивный | Поражает в основном мальчиков младшего возраста, аналогично СМА типа 1, но с диафрагмальный паралич |
Дистальная спинальная мышечная атрофия 2 типа (DSMA2)
| 605726 | SIGMAR1 | 19p13.3 | Аутосомно-рецессивный | Медленно прогрессирующий | |
(DSMA3)
| 607088 | ? | 11q13.3 | Аутосомно-рецессивный | Медленно прогрессивный | |
| (DSMA4) | 611067 | PLEKHG5 | 1p36.31 | Аутосомно рецессивный | Медленно прогрессирующий, описан только в одной семье | |
| (DSMA5) | 614881 | DNAJB2 | 2q35 | Аутосомно-рецессивный | Начало у молодых взрослых, медленно прогрессирующее | |
(DSMAVA)
| 600794 | GARS | 7p14.3 | Аутосомно-доминантная | с преобладанием верхних конечностей; аллельный и перекрывается с CMT2D, фенотип перекрывается с синдромом Сильвера | |
(DSMAVB)
| 614751 | REEP1 | 2p11 | Аутосомно-доминантный | С преобладанием верхних конечностей; аллельный и перекрывающийся с -31 | |
| 615575 | FBXO38 | 5q32 | Аутосомно-доминантный | Ювенильное или взрослое начало, медленно прогрессирующее, влияет на оба проксимальные и дистальные мышцы, первоначально проявляется слабостью икр, которая переходит в руки | |
| 158580 | SLC5A7 | 2q12.3 | Аутосомно-доминантный | Начало у взрослых с параличом голосовых связок, очень редко | |
Врожденная дистальная спинальная мышечная атрофия
| 600175 | TRPV4 | 12q24.11 | Аутосомно-доминантный | Поражает в первую очередь дистальные мышцы нижних конечностей, непрогрессирующий, редко, аллельный с и CMT2C | |
(SPSMA)
| 181405 | TRPV4 | 12q24.11 | Аутосомно-доминантный. или Х-сцепленный доминант | Поражает мышцы нижних конечностей, непрогрессирующее, редко, аллельное с врожденной дистой l спинальная мышечная атрофия и CMT2C | |
| 158590 | HSPB8 | 12q24.23 | Аутосомно-доминантная | Начало у взрослых. Аллельный с болезнью Шарко – Мари – Тута тип 2L (CMT2L) | |
| 182960 | ? | 7q34 – q36 | Аутосомно-доминантный | Ювенильный -начало | |
| (JSSMA) | 183020 | ? | 18q21.3 | ? | Ювенильное начало, прогрессирующее со стабилизацией через 2–4 года, поражает преимущественно руки, очень редко | |
| (SMA-FK) | 182980 | ВАПБ | 20q13.32 | Аутосомно-доминантный | Позднее начало, поражает проксимальные мышцы у взрослых | |
| Спинальная мышечная атрофия Джокелинского типа (SMA-J) | 615048 | CHCHD10 | 22q11.2 – q13.2 | Аутосомно-доминантный | Поздний, медленно прогрессирующий, поражает как проксимальные, так и дистальные мышцы у взрослых | |
| (SMALED1) | 158600 | DYNC1H1 | 14q32 | Аутосомно-доминантный | Поражает проксимальные мышцы у младенцев | |
| (SMALED2) | 615290 | BICD2 | 9q22.31 | Аутосомно-доминантный | Врожденный или ранний- начало, поражающее преимущественно нижние конечности, непрогрессирующее, очень редко | |
| Спинальная мышечная атрофия с прогрессирующей миоклонической эпилепсией (СМА -PME) | 159950 | ASAH1 | 8p22 | Аутосомно-рецессивный | ||
| (SMABF2) | 271225 | ASCC1 | 10q21 | Аутосомно-рецессивный | Характеризуется тяжелой мышечной массой истощение, как при СМА типа I, сопровождающееся врожденными переломами костей | |
| ПКГ | Спинальная мышечная атрофия с понтоцеребеллярной гипоплазией (SMA-PCH)
| 607596 | VRK1 | 14q32 | Аутосомно-доминантный | → см. Понтоцеребеллярная гипоплазия |
| ММА | Ювенильная асимметричная сегментарная спинальная мышечная атрофия (JASSMA)
| 602440 | ? | ? | ? | → см. Мономельная амиотрофия |
| PMA | Прогрессирующая мышечная атрофия позвоночника
| ? | ? | ? | ? | → см. Прогрессирующая мышечная атрофия |
При всех формах СМА (за исключением Х-сцепленной спинальной мышечной атрофии 1 типа ) только мотонейроны, расположенные в передний рог спинного мозга, поражены Тед; сенсорные нейроны, расположенные в заднем роге спинного мозга, не затрагиваются. Напротив, наследственные заболевания, которые вызывают как слабость из-за моторной денервации, так и сенсорное нарушение из-за сенсорной денервации, известны как (HMSN).
| Классификация | D |
|---|