
Войти
| Классификация | Эволюционная биология |
|---|---|
| Подклассификация | Молекулярная филогенетика |
| Критерии оптимального поиска | Байесовский вывод |
Байесовский вывод филогении использует функцию правдоподобия для создания величины, называемой апостериорная вероятность деревьев с использованием модели эволюции, основанной на некоторых априорных вероятностях, что дает наиболее вероятное филогенетическое дерево для заданных данных. Байесовский подход стал популярным благодаря прогрессу в скорости вычислений и интеграции алгоритмов цепи Маркова Монте-Карло (MCMC). Байесовский вывод имеет ряд применений в молекулярной филогенетике и систематике.
 Теорема Байеса
Теорема Байеса  Метафора, иллюстрирующая шаги метода MCMC
Метафора, иллюстрирующая шаги метода MCMC Байесовский Вывод относится к вероятностному методу, разработанному преподобным Томасом Байесом на основе теоремы Байеса. Опубликованный посмертно в 1763 году, он был первым выражением обратной вероятности и основой байесовского вывода. Независимо, не зная о работе Байеса, Пьер-Симон Лаплас разработал теорему Байеса в 1774 году.
Байесовский вывод широко использовался до 1900-х годов, когда произошел переход к частотному выводу, в основном из-за вычислительных ограничений. Основанный на теореме Байеса, байесовский подход объединяет априорную вероятность дерева P (A) с вероятностью данных (B) для получения апостериорного распределения вероятностей на деревьях P (A | B). Апостериорная вероятность дерева будет указывать на вероятность того, что дерево будет правильным, поскольку дерево с наивысшей апостериорной вероятностью выбрано для наилучшего представления филогении. Именно внедрение методов Монте-Карло цепи Маркова (MCMC) Николасом Метрополисом в 1953 году произвело революцию в байесовском выводе и к 1990-м годам стало широко используемым методом среди филогенетиков. Некоторыми из преимуществ перед традиционными методами максимальной экономии и максимального правдоподобия являются возможность учета филогенетической неопределенности, использование априорной информации и включение сложных моделей эволюции, которые ограничивали вычислительный анализ для традиционные методы. Несмотря на преодоление сложных аналитических операций, апостериорная вероятность по-прежнему включает суммирование по всем деревьям и, для каждого дерева, интегрирование по всем возможным комбинациям значений параметров модели замещения и длин ветвей.
Методы MCMC могут быть описаны в три этапа: сначала с использованием стохастического механизма предлагается новое состояние для цепи Маркова. Во-вторых, вычисляется вероятность того, что это новое состояние будет правильным. В-третьих, предлагается новая случайная величина (0,1). Если это новое значение меньше вероятности принятия, новое состояние принимается и состояние цепочки обновляется. Этот процесс выполняется тысячи или миллионы раз. Количество посещений одного дерева в ходе цепочки является всего лишь допустимым приближением его апостериорной вероятности. Некоторые из наиболее распространенных алгоритмов, используемых в методах MCMC, включают алгоритмы Metropolis-Hastings, Metropolis-Coupling MCMC (MC³) и LOCAL алгоритм Ларгета и Саймона.
Одним из наиболее распространенных используемых методов MCMC является алгоритм Метрополиса-Гастингса, модифицированная версия исходного алгоритма Метрополиса. Это широко используемый метод случайной выборки из сложных и многомерных вероятностей распределения. Алгоритм Метрополиса описывается следующими шагами:
Алгоритм продолжает работать, пока не достигнет равновесное распределение. Также предполагается, что вероятность предложения нового дерева T j, когда мы находимся в состоянии старого дерева T i, является такой же вероятностью предложения T i, когда мы находимся в T j. Когда это не так, применяются поправки Гастингса. Целью алгоритма Метрополиса-Гастингса является создание набора состояний с помощью det до тех пор, пока марковский процесс не достигнет стационарного распределения. Алгоритм состоит из двух компонентов:
Алгоритм MCMC, связанный с мегаполисом (MC³), был предложен для решения практической проблемы цепи Маркова, движущейся по пикам, когда целевое распределение имеет несколько локальных пиков, разделенных низкими впадинами, которые, как известно, существуют в пространстве дерева. Это имеет место во время эвристического поиска в дереве в соответствии с критериями максимальной экономии (MP), максимального правдоподобия (ML) и минимального развития (ME), и то же самое можно ожидать для поиска в стохастическом дереве с использованием MCMC. Эта проблема приведет к неправильному приближению образцов к апостериорной плотности. (MC³) улучшает перемешивание цепей Маркова при наличии нескольких локальных пиков в апостериорной плотности. Он запускает несколько (m) цепочек параллельно, каждая для n итераций и с разными стационарными распределениями 




![{\displaystyle \pi _{j}(\theta)=\pi (\theta)^{1/[1+\lambda (j-1)]},\ \ \lambda>0,}]( https://wikimedia.org/api/rest_v1/media/math/render/svg/2e9d8c159f93b004aa99ba2ce2249578daf6db1e )
, так что первая цепочка является холодовой цепью с правильная целевая плотность, тогда как цепочки 







В конце цикла используется вывод только из холодовой цепи, тогда как из горячих цепочек отбрасывается. Эвристически горячие цепи довольно легко посещают локальные пики, а переключение состояний между цепями позволит иногда холодной цепочке перепрыгивать впадины, что приводит к лучшему перемешиванию. Однако если 
Очевидным недостатком алгоритма является то, что 

ЛОКАЛЬНЫЙ алгоритм предлагает вычислительное преимущество по сравнению с предыдущими методами и демонстрирует, что байесовский подход может оценивать неопределенность с вычислительной точки зрения на больших деревьях. Алгоритм LOCAL является усовершенствованием алгоритма GLOBAL, представленного в Mau, Newton and Larget (1999), в котором все длины ветвей меняются в каждом цикле. ЛОКАЛЬНЫЕ алгоритмы изменяют дерево путем случайного выбора внутренней ветви дерева. Каждый узел на концах этой ветви связан с двумя другими ветвями. По одному из каждой пары выбирается случайным образом. Представьте, что вы берете эти три выбранных края и натягиваете их как веревку для белья слева направо, причем направление (влево / вправо) также выбирается случайным образом. Две конечные точки первой выбранной ветви будут иметь поддерево, висящее как кусок одежды, привязанный к линии. Алгоритм выполняется путем умножения трех выбранных ветвей на обычную случайную величину, что похоже на растяжение или сжатие бельевой веревки. Наконец, крайнее левое из двух подвешенных поддеревьев отсоединяется и снова прикрепляется к веревке для белья в месте, выбранном равномерно случайным образом. Это будет дерево кандидатов.
Предположим, мы начали с выбора внутренней ветви длиной 









Чтобы оценить длину ветви 





для неизменных сайтов и

Таким образом, ненормализованное апостериорное распределение:

или, альтернативно,

Обновить длину ветви, выбирая новое значение равномерно случайным образом из окна половинной ширины 

где 


Пример: 





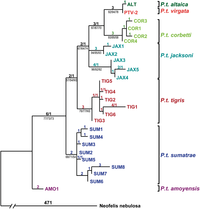 Филогенетические отношения тигра, значения начальной загрузки показаны в ветвях.
Филогенетические отношения тигра, значения начальной загрузки показаны в ветвях.  Пример притяжения длинной ветви. Более длинные ветви (A и C) кажутся более близкими.
Пример притяжения длинной ветви. Более длинные ветви (A и C) кажутся более близкими. Существует множество подходов к реконструкции филогенетических деревьев, каждый со своими преимуществами и недостатками, и нет однозначного ответа на вопрос «какой метод является лучшим?». Максимальная экономия (MP) и максимальная вероятность (ML) - традиционные методы, широко используемые для оценки филогении, и оба используют символьную информацию напрямую, как это делают байесовские методы.
Maximum Parsimony восстанавливает одно или несколько оптимальных деревьев на основе матрицы дискретных признаков для определенной группы таксонов и не требует модели эволюционного изменения. MP дает наиболее простое объяснение заданному набору данных, реконструируя филогенетическое дерево, которое включает в себя как можно меньше изменений в последовательностях; это то, которое демонстрирует наименьшее количество эволюционных шагов для объяснения взаимосвязи между таксонами. Поддержка ветвей дерева представлена процентом начальной загрузки. По той же причине, по которой он широко использовался, из-за своей простоты, MP также подвергся критике и был отодвинут на задний план ML и байесовскими методами. MP имеет несколько проблем и ограничений. Как показал Фельзенштейн (1978), MP может быть статистически несовместимым, что означает, что по мере накопления все большего и большего количества данных (например, длины последовательности) результаты могут сходиться на неверном дереве и приводить к притяжению длинных ветвей, филогенетический феномен, при котором таксоны с длинными ветвями (многочисленные изменения состояния признаков), как правило, оказываются более близкими в филогенезе, чем они есть на самом деле. Что касается морфологических данных, недавние исследования моделирования показывают, что экономия может быть менее точной, чем деревья, построенные с использованием байесовских подходов, возможно, из-за чрезмерной точности, хотя это оспаривается. Исследования с использованием новых методов моделирования продемонстрировали, что различия между методами вывода являются результатом используемой стратегии поиска и метода консенсуса, а не используемой оптимизации.
Как и в случае максимальной экономии, максимальная вероятность будет оценивать альтернативные деревья. Однако он учитывает вероятность того, что каждое дерево объясняет данные на основе модели эволюции. В этом случае дерево с наибольшей вероятностью объяснения данных выбирается среди других. Другими словами, он сравнивает, как разные деревья предсказывают наблюдаемые данные. Введение модели эволюции в анализ ML представляет собой преимущество перед MP, поскольку учитываются вероятность нуклеотидных замен и скорость этих замен, что более реалистично объясняет филогенетические отношения таксонов. Важным аспектом этого метода является длина ветвей, которую экономия игнорирует, поскольку изменения более вероятны на длинных ветвях, чем на коротких. Такой подход может устранить притяжение длинных ветвей и объяснить большую согласованность ML по сравнению с MP. Хотя многие считают ML лучшим подходом к выводу филогении с теоретической точки зрения, ML требует больших вычислительных ресурсов, и практически невозможно исследовать все деревья, поскольку их слишком много. Байесовский вывод также включает в себя модель эволюции, и его основные преимущества перед MP и ML заключаются в том, что он более эффективен с точки зрения вычислений, чем традиционные методы, он позволяет количественно определять и устранять источник неопределенности и может включать сложные модели эволюции.
MrBayes - это бесплатный программный инструмент, который выполняет байесовский вывод филогении. Первоначально написано Джоном П. Хуэльсенбеком и Фредериком Ронквистом в 2001 году. По мере роста популярности байесовских методов MrBayes стал одним из программ, предпочитаемых многими молекулярными филогенетиками. Он предлагается для операционных систем Macintosh, Windows и UNIX и имеет интерфейс командной строки. Программа использует стандартный алгоритм MCMC, а также вариант MCMC, связанный с Metropolis. MrBayes считывает выровненные матрицы последовательностей (ДНК или аминокислот) в стандартном формате NEXUS.
MrBayes использует MCMC для аппроксимации апостериорных вероятностей деревьев. Пользователь может изменить предположения модели замещения, априорные значения и детали анализа MC³. Он также позволяет пользователю удалять и добавлять в анализ таксоны и символы. Программа использует самую стандартную модель замены ДНК, 4x4, также называемую JC69, которая предполагает, что изменения по нуклеотидам происходят с равной вероятностью. Он также реализует ряд моделей 20x20 замен аминокислот и кодоновых моделей замены ДНК. Он предлагает различные методы для ослабления предположения о равных скоростях замен по нуклеотидным сайтам. MrBayes также может делать выводы о наследственных состояниях с учетом неопределенности филогенетического дерева и параметров модели.
MrBayes 3 был полностью реорганизованной и реструктурированной версией оригинального MrBayes. Основным нововведением была способность программного обеспечения учитывать неоднородность наборов данных. Эта новая структура позволяет пользователю смешивать модели и использовать преимущества эффективности байесовского анализа MCMC при работе с данными различного типа (например, белками, нуклеотидами и морфологическими данными). По умолчанию он использует Metropolis-Coupling MCMC.
MrBayes 3.2 Новая версия MrBayes была выпущена в 2012 г. Новая версия позволяет пользователям выполнять несколько анализов параллельно. Он также обеспечивает более быстрые вычисления вероятности и позволяет делегировать эти вычисления узлам обработки графики (GPU). Версия 3.2 предоставляет более широкие возможности вывода, совместимые с FigTree и другими программами просмотра деревьев.
В эту таблицу включены некоторые из наиболее распространенных филогенетических программ, используемых для определения филогенеза в рамках байесовской системы. Некоторые из них не используют исключительно байесовские методы.
| Имя | Описание | Метод | Автор | Ссылка на веб-сайт |
|---|---|---|---|---|
| Armadillo Workflow Platform | Workflow Platform, посвященная филогенетическим и общий биоинформатический анализ | Вывод филогенетических деревьев с использованием расстояний, максимального правдоподобия, максимальной экономичности, байесовских методов и связанных рабочих процессов | E. Лорд, М. Леклерк, А. Бок, А.Б. Диалло и В. Макаренковы | https://web.archive.org/web/20161024081942/http://www.bioinfo.uqam.ca/armadillo/. |
| Бали-Пхи | Одновременный байесовский вывод выравнивания и филогении | Байесовский вывод, выравнивание, а также поиск по дереву | Suchard MA, Redelings BD | http://www.bali-phy.org |
| BATWING | Байесовский анализ деревьев с генерацией внутренних узлов | Байесовский вывод, демографическая история, разделение населения | I. Дж. Уилсон, Д. Уил, Д. Болдинг | http://www.maths.abdn.ac.uk/˜ijw |
| Байесовская филогения | Байесовский вывод деревьев с использованием цепей Маркова Монте-Карло методы | Байесовский вывод, множественные модели, смешанная модель (автоматическое разбиение) | M. Пагель, А. Мид | http://www.evolution.rdg.ac.uk/BayesPhy.html |
| PhyloBayes / PhyloBayes MPI | Сэмплер байесовской цепи Монте-Карло Маркова (MCMC) для филогенетической реконструкции. | Непараметрические методы моделирования межсайтовых вариаций нуклеотидной или аминокислотной предрасположенности. | N. Лартиллот, Н. Родриг, Д. Стаббс, Дж. Ричер | http://www.atgc-montpellier.fr/phylobayes/ |
| BEAST | Байесовский эволюционный анализ деревьев выборки | Байесовский вывод, расслабленные молекулярные часы, демографическая история | А. Дж. Драммонд, А. Рамбаут и М.А. Сушард | https://beast.community |
| BEAST 2 | Программная платформа для байесовского эволюционного анализа | Байесовский вывод, пакеты, несколько моделей | R Bouckaert, J Heled, D Kühnert, T. Vaughan, CH Wu, D Xie, MA Suchard, A Rambaut, AJ Drummond. | http: //www.beast2.org |
| BUCKy | Байесовское соответствие деревьев генов | Байесовское согласование с использованием модифицированного жадного консенсуса некорневых квартетов | C. Ане, Б. Ларже, Д.А. Баум, С. Смит, А. Рокас, Б. Ларгет, С.К. Kotha, C.N. Дьюи, К. Ане | http://www.stat.wisc.edu/~ane/bucky/ |
| Geneious (плагин MrBayes) | Geneious предоставляет инструменты для исследования генома и протеома | Объединение соседей, UPGMA, плагин MrBayes, плагин PHYML, плагин RAxML, плагин FastTree, плагин GARLi, плагин PAUP * | A. J. Drummond, M.Suchard, V.Lefort et al. | http://www.geneious.com |
| MrBayes | Филогенетический вывод | Программа для байесовского вывода и выбор моделей из широкого диапазона филогенетических и эволюционных моделей. | Занг, Хюльсенбек, Дер Марк, Ронквист и Тесленко | https://nbisweden.github.io/MrBayes/ |
| TOPALi | Филогенетический вывод | Выбор филогенетической модели, байесовский анализ и оценка филогенетического древа максимального правдоподобия, обнаружение сайтов при положительном отборе и анализ местоположения контрольной точки рекомбинации | И.Милн, Д.Линднер, и др. | http://www.topali.org |
Байесовский вывод широко использовался молекулярными филогенетиками для широкого ряда приложений. Некоторые из них включают:
 Хронограмма, полученная в результате анализа молекулярных часов с использованием BEAST. Круговая диаграмма в каждом узле указывает возможные наследственные распределения, выведенные из байесовского двоичного анализа MCMC (BBM)
Хронограмма, полученная в результате анализа молекулярных часов с использованием BEAST. Круговая диаграмма в каждом узле указывает возможные наследственные распределения, выведенные из байесовского двоичного анализа MCMC (BBM)