
Войти
| Синдром Блау | |
|---|---|
| Другие названия | Артрокутаноувеальный гранулематоз |
 | |
| Грубые черты лица у мальчика с синдромом Блау | |
| Специальность | Дерматология |
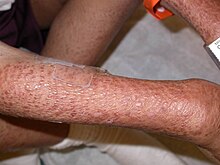 Множественные красновато-коричневые папулы, сливающиеся над правой рукой у мальчика с синдромом Блау
Множественные красновато-коричневые папулы, сливающиеся над правой рукой у мальчика с синдромом Блау Синдром Блау является аутосомным доминантным генетическим воспалительным заболеванием, поражающим кожу, глаза и суставы. Это вызвано мутацией в гене NOD2 (CARD15). Симптомы обычно появляются в возрасте до 4 лет, и заболевание проявляется в виде раннего кожного саркоидоза, гранулематозного артрита и увеита.
Выяснение того, что дефект гена в BS включает ген CARD15 / NOD2, имеет стимулировали многих исследователей, чтобы определить, как этот ген действует как часть врожденной иммунной системы, которая реагирует на бактериальные полисахариды, такие как мурамилдипептид, чтобы вызвать сигнальные пути, которые вызывают цитокиновые ответы и защищают организм. При BS генетический дефект, по-видимому, ведет к чрезмерной экспрессии и плохому контролю воспалительной реакции, что приводит к широко распространенному гранулематозу, воспалению и повреждению тканей.
Лечение включало обычные противовоспалительные препараты такие как надпочечники глюкокортикоиды, антиметаболиты, а также биологические агенты, такие как анти-TNF и инфликсимаб, все с различной степенью успеха.
В 1985 году Эдвард Блау, педиатр из Маршфилда, штат Висконсин, сообщил о семье, у которой более четырех поколений было гранулематозное воспаление кожи, глаз и суставов. Состояние было передано по аутосомно-доминантному признаку. В том же году Jabs et al. сообщили о семье, в которой более двух поколений страдали гранулематозным синовитом, увеитом и черепными невропатиями. Заболевание передалось по аутосомно-доминантному типу. В 1981 г. Malleson et al. сообщили о семье, у которой был аутосомно-доминантный синовит, камптодактилия и иридоциклит. Один участник умер от гранулематозного артериита сердца и аорты. В 1982 году Ротенштейн сообщил о семье с гранулематозным артериитом, сыпью, иритом и артритом, которые передавались как аутосомно-доминантный признак в течение трех поколений. Затем в 1990 году Pastores et al. сообщил о родственнике с фенотипом, очень похожим на то, что описал Блау, и предложил назвать это состояние синдромом Блау (БС). Они также указали на сходство в семьях, отмеченных выше, с BS, но также указали на значительные различия в фенотипах.
В 1996 г. Tromp et al. провели широкий поиск генома с использованием затронутых и незатронутых членов исходной семьи. Маркер, D16S298, дал максимальный балл LOD 3,75 и поместил локус восприимчивости BS в интервал 16p12-q21. Hugot et al. обнаружил локус восприимчивости к болезни Крона, гранулематозному воспалению кишечника, на хромосоме 16 рядом с локусом BS. На основании вышеизложенной информации Блау предположил в 1998 г., что генетический дефект БС и болезни Крона может быть одинаковым или подобным.
Наконец, в 2001 году Miceli-Richard et al. обнаружили, что дефект в BS находится в нуклеотид-связывающем домене CARD15 / NOD2. В своей статье они отметили, что мутации в CARD15 также были обнаружены при болезни Крона. Подтверждение наличия дефекта BS в гене CARD15 было сделано Wang et al. в 2002 году с использованием семейства BS и других. С этой информацией диагноз BS был определен не только по фенотипу, но теперь и по генотипу.
Раннее начало саркоидоз - это BS без семейного анамнеза, BS был диагностирован у пациентов, у которых есть не только классическая триада, но и гранулема во многих органах.
| Классификация | D
|
|---|---|
| Внешние ресурсы |