
Войти
 Использование секвенирования белков и пептидов Beckman-Spinco, 1970
Использование секвенирования белков и пептидов Beckman-Spinco, 1970 Секвенирование белков - это практический процесс определения аминокислотная последовательность всего или части белка или пептида. Это может служить для идентификации белка или характеристики его посттрансляционных модификаций. Как правило, частичное секвенирование белка предоставляет достаточно информации (один или несколько тегов последовательности) для его идентификации со ссылкой на базы данных последовательностей белков, полученных в результате концептуальной трансляции генов.
Два основных прямых методами секвенирования белков являются масс-спектрометрия и деградация по Эдману с использованием секвенатора белков (секвенатор). В настоящее время методы масс-спектрометрии наиболее широко используются для секвенирования и идентификации белков, но деградация по Эдману остается ценным инструментом для характеристики N-конца.
Это часто желательно знать неупорядоченный аминокислотный состав белка до попытки найти упорядоченную последовательность, так как эти знания можно использовать для облегчения обнаружения ошибок в процессе определения последовательности или для различения неоднозначных результатов. Знание частоты встречаемости определенных аминокислот также может быть использовано для выбора, какую протеазу использовать для переваривания белка. Также может быть определено неправильное включение низких уровней нестандартных аминокислот (например, норлейцина) в белки. Обобщенный метод, который часто называют аминокислотным анализом для определения частоты встречаемости аминокислот, заключается в следующем:
Гидролиз выполняется путем нагревания образца белка в 6 M соляной кислоте до 100–110 ° C в течение 24 часов или дольше. Белки с большим количеством объемных гидрофобных групп могут потребовать более длительных периодов нагревания. Однако эти условия настолько сильны, что некоторые аминокислоты (серин, треонин, тирозин, триптофан, глутамин и цистеин ) разлагаются. Чтобы обойти эту проблему, Biochemistry Online предлагает нагревать отдельные образцы в течение разного времени, анализировать каждый полученный раствор и экстраполировать обратно на нулевое время гидролиза. Расталл предлагает различные реагенты для предотвращения или уменьшения разложения, такие как тиол реагенты или фенол для защиты триптофана и тирозина от воздействия хлора и предварительного окисления. цистеин. Он также предлагает измерить количество выделившегося аммиака, чтобы определить степень гидролиза амида.
Аминокислоты можно разделить с помощью ионного обмена. хроматография затем дериватизировали, чтобы облегчить их обнаружение. Чаще всего аминокислоты дериватизируются, а затем разделяются с помощью ВЭЖХ с обращенной фазой.
Пример ионообменной хроматографии дается NTRC с использованием сульфированного полистирола в качестве матрицы, добавления аминокислот в кислотный раствор и прохождения буфер стабильно увеличивающегося pH через колонку. Аминокислоты элюируются, когда pH достигает их соответствующих изоэлектрических точек. После разделения аминокислот определяют их соответствующие количества, добавляя реагент, который образует окрашенное производное. Если количество аминокислот превышает 10 нмоль, для этого можно использовать нингидрин ; он дает желтый цвет при реакции с пролином и ярко-фиолетовый цвет с другими аминокислотами. Концентрация аминокислоты пропорциональна поглощению полученного раствора. При очень малых количествах, вплоть до 10 пмоль, флуоресцентные производные могут быть образованы с использованием таких реагентов, как орто-фтальдегид (OPA) или флуоресскамин.
. При дериватизации перед колонкой может использоваться реагент Эдмана для получения производная, обнаруживаемая УФ-светом. Более высокая чувствительность достигается при использовании реагента, образующего флуоресцентное производное. Дериватизированные аминокислоты подвергают обращенно-фазовой хроматографии, обычно с использованием колонки с диоксидом кремния C8 или C18 и оптимизированного градиента элюирования. Элюирующие аминокислоты обнаруживают с помощью УФ-детектора или флуоресцентного детектора, а площади пиков сравнивают с площадями для производных стандартов, чтобы количественно определить каждую аминокислоту в образце.
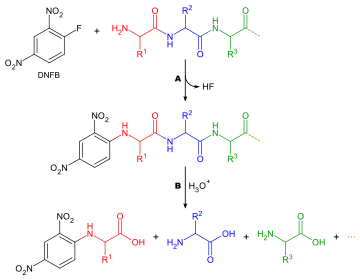 Метод Сэнгера анализа концевых пептидных групп: A дериватизация N-концевого конца с помощью реагента Сэнгера (DNFB), B общий кислотный гидролиз динитрофенилпептида
Метод Сэнгера анализа концевых пептидных групп: A дериватизация N-концевого конца с помощью реагента Сэнгера (DNFB), B общий кислотный гидролиз динитрофенилпептида Определение того, какая аминокислота образует N-конец цепи пептида, полезно по двум причинам: упорядочение последовательностей отдельных пептидных фрагментов в целую цепь, и потому что первый раунд деградации по Эдману часто загрязнен примесями и, следовательно, не дает точного определения N-концевой аминокислоты. Ниже приводится обобщенный метод анализа N-концевых аминокислот:
Существует множество различных реагентов, которые можно использовать для мечения концевых аминокислот. Все они реагируют с аминогруппами и, следовательно, также будут связываться с аминогруппами в боковых цепях аминокислот, таких как лизин - по этой причине необходимо быть осторожным при интерпретации хроматограмм, чтобы убедиться, что выбрано правильное пятно. Двумя наиболее распространенными реагентами являются реагент Сенгера (1-фтор-2,4-динитробензол ) и производные дансила, такие как дансилхлорид. Фенилизотиоцианат, реагент для разложения Эдмана, также может быть использован. Здесь применяются те же вопросы, что и при определении аминокислотного состава, за исключением того, что окрашивание не требуется, поскольку реагенты производят окрашенные производные, и требуется только качественный анализ. Таким образом, аминокислоту не нужно элюировать из хроматографической колонки, а просто сравнивать со стандартом. Еще одно соображение, которое следует принять во внимание, заключается в том, что, поскольку любые аминогруппы будут реагировать с реагентом для метки, нельзя использовать ионообменную хроматографию и тонкослойную хроматографию или жидкостную хроматографию высокого давления следует использовать вместо этого.
Количество доступных методов анализа C-концевых аминокислот намного меньше, чем количество доступных методов анализа N-концов. Наиболее распространенный метод - это добавление карбоксипептидаз к раствору белка, регулярный отбор образцов и определение конечной аминокислоты путем анализа графика зависимости концентраций аминокислот от времени. Этот метод будет очень полезен в случае полипептидов и N-концов, блокированных белком. С-концевое секвенирование может значительно помочь в проверке первичных структур белков, предсказанных на основе последовательностей ДНК, и для обнаружения пострансляционного процессинга генных продуктов из известных последовательностей кодонов.
деградация по Эдману является очень важной реакцией для секвенирования белка, поскольку она позволяет обнаружить упорядоченный аминокислотный состав белка. В настоящее время широко используются автоматические секвенаторы Эдмана, которые способны секвенировать пептиды длиной примерно до 50 аминокислот. Схема реакции для секвенирования белка деградацией Эдмана следует; некоторые из шагов будут подробно описаны позже.
Пептиды длиной более 50-70 аминокислот не могут быть надежно секвенированы с помощью деградации по Эдману. Из-за этого длинные белковые цепи необходимо разбивать на небольшие фрагменты, которые затем можно секвенировать индивидуально. Переваривание осуществляется либо эндопептидазами, такими как трипсин или пепсин, либо химическими реагентами, такими как бромид цианогена. Различные ферменты дают разные модели расщепления, и перекрытие между фрагментами можно использовать для построения общей последовательности.
Пептид, подлежащий секвенированию, адсорбируется на твердой поверхности. Одна распространенная подложка представляет собой стекловолокно, покрытое полибреном, катионным полимером. Реагент Эдмана, фенилизотиоцианат (PITC), добавляют к адсорбированному пептиду вместе со слабощелочным буферным раствором 12% триметиламина. Он реагирует с аминогруппой N-концевой аминокислоты.
Конечная аминокислота затем может быть избирательно отделена путем добавления безводной кислоты. Затем производное изомеризуется с образованием замещенного, которое можно смыть и идентифицировать с помощью хроматографии, и цикл можно повторить. Эффективность каждого этапа составляет около 98%, что позволяет надежно определить около 50 аминокислот.
 Аппарат для секвенирования белков Beckman-Coulter Porton LF3000G
Аппарат для секвенирования белков Beckman-Coulter Porton LF3000G A секвенатор протеинов - это аппарат, который выполняет деградацию по Эдману в автоматическом режиме. Образец белка или пептида иммобилизуют в реакционном сосуде секвенатора белка и проводят деградацию по Эдману. Каждый цикл высвобождает и дериватизирует одну аминокислоту с N-конца белка или пептида, а затем высвобожденное производное аминокислоты идентифицируется с помощью ВЭЖХ. Процесс секвенирования повторяется для всего полипептида до тех пор, пока не будет установлена вся измеряемая последовательность, или в течение заранее определенного количества циклов.
Идентификация белка - это процесс присвоения имени интересующему белку (POI) на основе его аминокислотной последовательности. Обычно только часть последовательности белка необходимо определить экспериментально, чтобы идентифицировать белок со ссылкой на базы данных последовательностей белков, выведенных из последовательностей ДНК их генов. Дальнейшая характеристика белка может включать подтверждение фактических N- и C-концов POI, определение вариантов последовательности и идентификацию любых присутствующих посттрансляционных модификаций.
Описана общая схема идентификации белка.
Образец последовательности фрагментация пептида позволяет напрямую определять его последовательность с помощью секвенирования de novo. Эта последовательность может использоваться для сопоставления баз данных последовательностей белков или для исследования посттрансляционных или химических модификаций. Это может предоставить дополнительные доказательства для идентификации белков, выполненной, как указано выше.
Пептиды, сопоставленные во время идентификации белка, не обязательно включают N- или C-концы, предсказанные для сопоставленного белка. Это может быть результатом того, что N- или C-концевые пептиды трудно идентифицировать с помощью MS (например, они слишком короткие или слишком длинные), посттрансляционно модифицированы (например, N-концевое ацетилирование) или действительно отличаются от предсказанных. Посттрансляционные модификации или усеченные концы могут быть идентифицированы путем более тщательного изучения данных (т.е. секвенирования de novo). Также может быть полезно повторное расщепление с использованием протеазы различной специфичности.
Хотя для определения посттрансляционных модификаций можно использовать подробное сравнение данных MS с прогнозами, основанными на известной последовательности белка, могут также использоваться целевые подходы к сбору данных. Например, специфическое обогащение фосфопептидами может помочь в идентификации сайтов фосфорилирования в белке. Альтернативные методы фрагментации пептидов в масс-спектрометре, такие как ETD или ECD, могут дать информацию о комплементарной последовательности.
Полная масса белка - это сумма масс его аминокислотных остатков плюс масса молекулы воды с поправкой на любые посттрансляционные модификации. Хотя белки ионизируются хуже, чем пептиды, полученные из них, белок в растворе может быть подвергнут ESI-MS и его масса измерена с точностью до 1 части из 20 000 или лучше. Этого часто бывает достаточно, чтобы подтвердить концы (таким образом, что измеренная масса белка совпадает с предсказанной по его последовательности) и сделать вывод о наличии или отсутствии многих посттрансляционных модификаций.
Протеолиз не всегда дает набор легко анализируемых пептидов, покрывающих всю последовательность POI. При фрагментации пептидов в масс-спектрометре часто не образуются ионы, соответствующие расщеплению каждой пептидной связи. Таким образом, выведенная последовательность для каждого пептида не обязательно является полной. Стандартные методы фрагментации не различают остатки лейцина и изолейцина, поскольку они изомерные.
Поскольку деградация по Эдману происходит от N-конца белка, это не сработает, если N-конец был химически модифицирован (например, путем ацетилирования или образования пироглутаминовой кислоты). Деградация по Эдману обычно не используется для определения положения дисульфидных мостиков. Также требуется количество пептида 1 пикомоль или выше для различимых результатов, что делает его менее чувствительным, чем масс-спектрометрия.
В биологии белки производятся посредством трансляции информационной РНК (мРНК) с белковой последовательностью, происходящей из последовательности кодонов в мРНК. Сама мРНК образуется в результате транскрипции генов и может быть дополнительно модифицирована. Эти процессы достаточно изучены, чтобы использовать компьютерные алгоритмы для автоматизации предсказаний последовательностей белков на основе последовательностей ДНК, например, из проектов секвенирования ДНК всего генома, и привели к созданию больших баз данных последовательностей белков, таких как UniProt. Предсказанные белковые последовательности являются важным ресурсом для идентификации белков с помощью масс-спектрометрии.
Исторически короткие последовательности белка (от 10 до 15 остатков), определенные деградацией по Эдману, были обратно транслированы в последовательности ДНК, которые можно было использовать в качестве зондов или праймеров для выделения молекулярных клонов соответствующего гена. или комплементарная ДНК. Затем определяли последовательность клонированной ДНК и использовали ее для определения полной аминокислотной последовательности белка.
Инструменты биоинформатики существуют для помощи в интерпретации масс-спектров (см. секвенирование пептидов De novo ), для сравнения или анализа последовательностей белков (см. Последовательность анализ ) или поиск в базах данных с использованием пептидных или белковых последовательностей (см. BLAST ).