
Войти
| Несовершенный остеогенез | |
|---|---|
| Другие названия | Хрупкая кость болезнь, синдром Лобштейна, fragilitas ossium, болезнь Вролика, остеопсатироз, болезнь Порака, болезнь Дюранте |
 | |
| Классические синие склеры человека с несовершенным остеогенезом | |
| Специальность | Педиатрия, медицинская генетика, остеология |
| Симптомы | Кости, которые легко ломаются,, синий оттенок белков глаз, небольшой рост, свободные суставы, потеря слуха |
| Продолжительность | Долгосрочная |
| Причины | Генетические (аутосомно-доминантные, новые мутация ) |
| Метод диагностики | На основании симптомов ДНК-тестирование |
| Лечение | Здоровый образ жизни (упражнения, отказ от курения), металлические стержни через длинные кости |
| Прогноз | Зависит от типа |
| Частота | 1 из 15 000 человек |
Остеогенез imperfecta (OI), также известная как болезнь хрупкости костей, представляет собой группу генетических нарушений, которые в основном поражают кости. В результате получаются кости, которые легко ломаются. Степень тяжести может быть от легкой до тяжелой. Другие симптомы могут включать синий оттенок белков глаз, небольшой рост, свободные суставы, потерю слуха, проблемы с дыханием и проблемы с зубами. Осложнения могут включать расслоение шейной артерии и расслоение аорты.
. Основным механизмом обычно является проблема соединительной ткани из-за нехватки коллагена I типа. Это происходит более чем в 90% случаев из-за мутаций в генах COL1A1 или COL1A2. Эти генетические проблемы часто возникают аутосомно-доминантно или возникают в результате новой мутации. Существует как минимум восемь основных типов, из которых тип I является наименее тяжелым, а тип II - наиболее тяжелым. Диагноз часто основывается на симптомах и может быть подтвержден тестом на коллаген или ДНК.
Нет лекарства. Поддержание здорового образа жизни с помощью физических упражнений и отказ от курения может помочь предотвратить переломы. Лечение может включать лечение переломов костей, обезболивающие, физиотерапию, использование скоб или инвалидных кресел и хирургическое вмешательство. Для их укрепления может быть проведена операция, при которой металлические стержни вводятся в длинные кости. Предварительные данные подтверждают использование лекарств типа бисфосфонат.
ОИ поражает примерно одного человека из 15 000. Исходы зависят от типа заболевания. Однако у большинства людей результаты хорошие. Состояние описывается с древней истории. Термин «несовершенный остеогенез» вошел в употребление в 1895 году и означает несовершенное формирование костей.
ОИ вызывает очень жидкую кровь сосудов, а также может привести к легким ушибам людей. Ослабление мышц приведет к деформации костей и проблемам с ростом.
Около 50% взрослых с НО испытывают значительную потерю слуха, часто более раннюю по сравнению с основное население. Потеря слуха при НО может быть связана или не связана с видимыми деформациями косточек и внутреннего уха. Потеря слуха часто начинается между вторым и четвертым десятилетием жизни и может быть кондуктивной, нейросенсорной или смешанной по своей природе.
НО ассоциируется с рядом неврологических аномалий, обычно связанных с центральная нервная система из-за деформации окружающих ее скелетных структур. Неврологические осложнения могут отрицательно повлиять на ожидаемую продолжительность жизни, и для коррекции тяжелых осложнений может потребоваться нейрохирургическое вмешательство.
НО может быть связано с повторяющимися болями в животе и хроническим запором, согласно двум исследованиям на предметах подвержены OI.
Существует не менее девяти различных типов OI. Тип I - самый распространенный. Симптомы варьируются от человека к человеку.
| Тип | Описание | Ген | OMIM | Тип наследования | Заболеваемость |
| I | легкая | Нулевой COL1A1 аллель | 166200 | аутосомно-доминантный, 60% de novo | 1 из 30,000 |
| II | тяжелая и обычно летальная в перинатальном периоде | COL1A1, COL1A2, | 166210 (IIA), 610854 (IIB) | аутосомно-доминантный, ~ 100% de novo | 1 из 40,000 до 1 из 100,000 |
| III | считается прогрессирующая и деформирующаяся | COL1A1, COL1A2 | 259420 | аутосомно-доминантная, ~ 100% de novo | 1 из 60,000 |
| IV | деформируется, но с нормальными склер в большинстве случаев | COL1A1, COL1A2 | 166220 | аутосомно-доминантный, 60% de novo | |
| V | имеет те же клинические признаки IV, но имеет уникальные гистологические данные («сетчатый») | IFITM5 | 610967 | аутосомно-доминантный | |
| VI | имеет те же клинические признаки IV, но имеет уникальные гистологические результаты («рыбья чешуя») | SERPINF1 | 610968 | аутосомно-рецессивный | |
| VII | ассоциированный с хрящ-ассоциированным белком | CRTAP | 610682 | аутосомно-рецессивным | |
| VIII | тяжелым или летальным исходом, связанный с белком лепрекан | LEPRE1, | 610915 | аутосомно-рецессивный | |
| IX | PPIB | аутосомно-рецессивный |
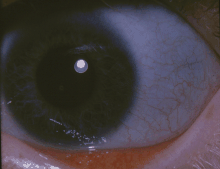 Голубая склера при несовершенном остеогенезе
Голубая склера при несовершенном остеогенезе Коллаген нормального качества, но вырабатывается в недостаточных количествах.
IA и IB определяется как отсутствие / присутствие несовершенного дентиногенеза (характеризуется опалесцирующими зубами; отсутствует в IA, присутствует в IB). Ожидаемая продолжительность жизни немного снижена по сравнению с населением в целом из-за возможности смертельных переломов костей и осложнений, связанных с ОИ I типа, таких как базилярная инвагинация.
Коллаген недостаточного качества или количество.
Тип II может быть далее подразделен на группы A, B и C, которые различаются рентгенологическим исследованием длинных костей и ребер. Тип IIA демонстрирует широкие и короткие длинные кости с широкими ребрами с ребрами. Тип IIB демонстрирует широкие и короткие длинные кости с тонкими ребрами, которые практически не имеют выступов. Тип IIC демонстрирует тонкие и длинные длинные кости с тонкими ребрами и ребрами с бусинами.
Коллаген сформирован неправильно, вырабатывается достаточное количество коллагена, но он дефектный.
Тип III выделяется среди других классификаций как «прогрессирующий деформирующий» тип, при котором новорожденный проявляет легкие симптомы при рождении и развивает вышеупомянутые симптомы на протяжении всего жизнь. Продолжительность жизни может быть нормальной, хотя и с серьезными физическими недостатками.
Коллаген достаточен, но не хорошего качества
Подобно типу I, тип IV может быть далее подразделен на типы IVA и IVB, характеризующиеся отсутствием ( IVA) или наличие (IVB) несовершенного дентиногенеза.
 Рентгеновский снимок правой ноги у новорожденного с НО типа V. Есть несколько деформированные длинные кости (в основном бедренная кость ) с расширенными метафизами. Имеется кортикальный перелом малоберцовой кости.
Рентгеновский снимок правой ноги у новорожденного с НО типа V. Есть несколько деформированные длинные кости (в основном бедренная кость ) с расширенными метафизами. Имеется кортикальный перелом малоберцовой кости.. Имея те же клинические признаки, что и тип IV, он гистологически отличается по внешнему виду "сетчатой" кости. Далее характеризуется «V-триадой», состоящей из (а) рентгеноконтрастной полосы, прилегающей к пластинам роста, (б) гипертрофических мозолей в местах перелома, и (в) кальцификации радио - локтевой кости. межкостная перепонка.
OI Тип V приводит к кальцификации перепонки между двумя костями предплечья, что затрудняет поворот запястья. Другой симптом - аномально большое количество восстанавливаемой ткани (гиперплазическая костная мозоль) в месте перелома. Другие особенности этого состояния включают вывих головки лучевой кости, искривление длинных костей и смешанную потерю слуха.
По крайней мере некоторые случаи этого типа вызваны мутациями в гене IFITM5.

OI Тип V у взрослого

OI Тип V у ребенка
Обладая теми же клиническими признаками, что и тип IV, он гистологически различается по внешнему виду костей «рыбьей чешуи». Недавно было обнаружено, что тип VI вызывается мутацией потери функции в гене SERPINF1. SERPINF1, член семейства серпиновых, также известен как фактор, производный от пигментного эпителия (PEDF), самый мощный эндогенный антиангиогенный фактор у млекопитающих.
В 2006 году была обнаружена рецессивная форма, названная «Тип VII» (фенотип от тяжелого до летального). Пока что это, кажется, ограничено первыми нациями людьми в Квебеке. Этот тип вызывает мутации в гене CRTAP.
OI, вызванный мутацией в гене LEPRE1, классифицируется как тип VIII.
Тип IX
Несовершенный остеогенез типа IX (OI9) вызван гомозиготной или сложной гетерозиготной мутацией в гене PPIB на хромосоме 15q22.
Тип X
Вызван гомозиготной мутацией в гене SERPINH на хромосоме 11q13.
OI, вызванный мутациями в FKBP10 на хромосоме 17q21. Мутации вызывают снижение секреции тримерных молекул проколлагена. Эти мутации также могут вызывать аутосомно-рецессивный синдром Брука, похожий на НО.
OI, вызванный мутацией сдвига рамки считывания в SP7. Эта мутация вызывает деформации костей, переломы и задержку прорезывания зубов.
OI, вызванный мутацией в гене костного морфогенного белка 1 (BMP1). Эта мутация вызывает рецидивирующие переломы, высокую костную массу и гиперэкстензивные суставы.
OI, вызванный мутациями в гене TMEM38B. Эта мутация вызывает повторные переломы и остеопению.
OI, вызванный гомозиготными или составными гетерозиготными мутациями в гене WNT1 на хромосоме 12q13. Это аутосомно-рецессивный тип.
OI, вызванный мутациями в гене CREB3L1. Эта мутация вызывает пренатальное начало повторных переломов ребер и длинных костей, деминерализацию, снижение оссификации черепа и синих склер. Члены семьи, гетерозиготные по OI XVI, могут иметь рецидивирующие переломы, остеопению и синие склеры.
OI, вызванный гомозиготной мутацией в гене SPARC на хромосоме 5q33.
. Тип XVIII
OI, вызванный гомозиготной мутацией в гене FAM46A на хромосоме 6q14. Характеризуется врожденным искривлением длинных костей, червячных костей, синих склер, коллапсом позвонков и множественными переломами в первые годы жизни.
Сообщалось о семье с несовершенным рецессивным остеогенезом иметь мутацию в гене хромосомы 9. Этот ген кодирует TRIC-B, компонент TRIC, моновалентный катион -специфический канал, участвующий в высвобождении кальция из внутриклеточных запасов.
Весьма вероятно, что есть другие гены, связанные с этим заболеванием, о которых еще не сообщалось.
Несовершенный остеогенез - это генетическое заболевание, которое вызывает повышенное количество переломов костей и дефекты коллагена. Основные причины развития заболевания являются результатом мутаций в генах COL1A1 и COL1A2, которые отвечают за выработку коллагена типа 1. Примерно 90% людей с НО являются гетерозиготными по мутациям в генах COL1A1 и COL1A2. Существует несколько факторов, которые являются результатом доминирующей формы расстройства НО. Эти факторы включают: внутриклеточный стресс, аномальная минерализация ткани, аномальные межклеточные взаимодействия, аномальные взаимодействия между клетками, нарушенная структура клеточного матрикса и нарушения между неколлагеновыми белками и коллагеном. Предыдущие исследования привели к убеждению, что ОИ является аутосомно-доминантным заболеванием с несколькими другими вариациями в геномах. Однако в последние несколько лет были выявлены аутосомно-рецессивные формы заболевания. Рецессивные формы OI в значительной степени связаны с дефектами шаперонов коллагена, ответственных за выработку проколлагена и сборку родственных белков. Примеры коллагеновых шаперонов, которые являются дефектными у пациентов с ОИ, включают шаперон HSP47 (синдром Коула-Карпентера ) и FKBP65. Мутации в этих шаперонах приводят к неправильной структуре сворачивания белков коллагена 1, что вызывает рецессивную форму нарушения. Существует три важных типа OI, которые являются результатом мутаций в комплексе пролил-3-гидроксилирования коллагена (компоненты CRTAP, P3H1 и CyPB). Эти компоненты отвечают за модификацию коллагена a1 (l) Pro986. Мутации в других генах, таких как SP7, SERPINF1, TMEM38B и BMP1, также могут приводить к нерегулярно сформированным белкам и ферментам, что приводит к рецессивной форме несовершенного остеогенеза. В настоящее время существуют связи с дефектами в других белках, вызванными генетическими мутациями, функция которых варьируется от структурных белков до ферментативных белков. Связь между белками, такими как фактор, производный пигментного эпителия (PEDF) и ограниченный костью интерферон-индуцированный трансмембранный белок (BRIL), является причиной несовершенного остеогенеза типов V и VI. Дефекты этих белков приводят к нарушению минерализации костей, что способствует формированию симптома хрупкости костей при несовершенном остеогенезе. Кроме того, мутации в генах COL1A1 и COL1A2 могут приводить к нарушениям передачи сигналов внеклеточного матрикса, который присутствует в белках коллагена, вызывая ухудшение симптомов заболевания. Одноточечная мутация в нетранслируемой 5'-области гена IFITM5 была недавно обнаружена и напрямую связана с OI типа V. Еще одна точечная мутация в области, которая кодирует белки коллагена в гене IFITM5, также была обнаружена у пациентов с существенно более тяжелые версии НО, чем просто тип V. Несовершенный остеогенез в некоторых редких случаях рассматривается как генетическое нарушение, связанное с Х-хромосомой, но продолжает оставаться преимущественно гетерозиготным доминантным заболеванием.
Люди с НО рождаются с дефектной соединительной тканью или неспособны вырабатывать ее, обычно из-за дефицита коллагена типа I. Этот дефицит возникает из-за замены аминокислоты глицина на более объемные аминокислоты в структуре коллагена тройной спирали. Более крупные боковые цепи аминокислот создают стерические препятствия, которые создают выпуклость в коллагеновом комплексе, что, в свою очередь, влияет как на молекулярную наномеханику, так и на взаимодействие между молекулами, которые находятся под угрозой. В результате организм может отреагировать гидролизом неправильной структуры коллагена. Если организм не разрушает неподходящий коллаген, соотношение между фибриллами коллагена и кристаллами гидроксиапатита, образующими кость, изменяется, вызывая хрупкость. Другой предполагаемый механизм заболевания заключается в том, что стрессовое состояние внутри коллагеновых фибрилл изменяется в местах мутаций, где локально более высокие силы сдвига приводят к быстрому разрушению фибрилл даже при умеренных нагрузках, поскольку гомогенное стрессовое состояние, обнаруживаемое в здоровых коллагеновых фибриллах, теряется. Эти недавние работы предполагают, что ОИ следует понимать как многомасштабное явление, которое включает механизмы на генетическом, нано-, микро- и макроуровне тканей. Большинство людей с НО получают его от родителей, но в 35% случаев это индивидуальная (de novo или «спорадическая») мутация.
Диагноз обычно основан на медицинской визуализации, включая обычные рентгеновские лучи и симптомы. Признаки на медицинских изображениях включают аномалии во всех концах и позвоночнике. Диагноз ОИ может быть подтвержден с помощью анализа ДНК или коллагена, но во многих случаях для постановки диагноза достаточно наличия переломов костей с небольшой травмой и наличия других клинических признаков, таких как синяя склера. Биопсия кожи может быть выполнена для определения структуры и количества коллагена I типа. Тестирование ДНК может подтвердить диагноз, однако не может исключить его, потому что не все мутации, вызывающие ОИ, известны и / или проверены. ОИ II типа часто диагностируется с помощью УЗИ во время беременности, когда уже могут присутствовать множественные переломы и другие характерные особенности. По сравнению с контролем, OI кортикальной кости показывает повышенную пористость, диаметр канала и связность при микрокомпьютерной томографии. Тяжелые типы ОИ обычно можно выявить до рождения с помощью метода генетического тестирования in vitro.
Чтобы определить, присутствует ли несовершенный остеогенез, генетическое секвенирование COL1A1, Могут быть использованы гены COL1A2 и IFITM5. Тестирование на дублирование и удаление также рекомендуется родителям, подозревающим, что у их ребенка НО. Наличие мутаций сдвига рамки считывания, вызванных дупликациями и делециями, обычно является причиной повышенной тяжести заболевания.
Важным дифференциальным диагнозом ОИ является жестокое обращение с детьми, поскольку оба могут иметь множественные переломы на разных стадиях заживления. Дифференцировать их может быть сложно, особенно при отсутствии других характерных признаков ОИ. Другие дифференциальные диагнозы включают рахит, остеомаляцию и другие редкие скелетные синдромы.
Нет лекарства. Поддержание здорового образа жизни с помощью физических упражнений и отказ от курения может помочь предотвратить переломы. Лечение может включать лечение сломанных костей, обезболивающие, физиотерапию, использование скоб или инвалидных колясок и хирургическое вмешательство. Для их укрепления может быть применен тип хирургического вмешательства, при котором металлические стержни вводятся в длинные кости.
Инфекции костей лечатся по мере их возникновения с помощью соответствующих антибиотики и антисептики.
В 1998 году клиническое испытание продемонстрировало эффективность внутривенного памидроната, бисфосфоната, который ранее использовался у взрослых для лечения остеопороза. При тяжелой ОИ памидронат уменьшил боль в костях, предотвратил новые переломы позвонков, изменил форму тел позвонков с ранее сломанными телами и уменьшил количество переломов длинных костей.
Хотя пероральные бисфосфонаты более удобны и дешевле, они не всасываются в виде ну, а внутривенные бисфосфонаты обычно более эффективны, хотя это изучается. В некоторых исследованиях были обнаружены бисфосфонаты для перорального и внутривенного введения, такие как пероральный алендронат и эквивалент памидроната для внутривенного введения. В исследовании с участием детей с легкой формой ОИ пероральный прием ризедроната увеличивал минеральную плотность костной ткани и уменьшал количество непозвоночных переломов. Однако это не привело к уменьшению количества новых переломов позвонков. В Кокрановском обзоре в 2016 году сделан вывод о том, что, хотя бисфосфонаты, похоже, улучшают минеральную плотность костей, неясно, приводит ли это к уменьшению числа переломов или улучшению качества жизни людей с несовершенным остеогенезом.
Бисфосфонаты менее эффективны при ОИ у взрослых.
Металлические стержни могут быть хирургически вставлены в длинные кости для повышения прочности, процедура, разработанная Гарольдом А. Софилд в Детских больницах Шрайнерс в Чикаго. В конце 1940-х годов Софилд, начальник штаба больниц Шрайнерс в Чикаго, работал там с большим количеством детей с ОИ и экспериментировал с различными методами укрепления костей у этих детей. В 1959 году Софилд вместе с Эдвардом А. Миллером написал основополагающую статью, в которой описал решение, которое в то время казалось радикальным: введение стержней из нержавеющей стали в интрамедуллярные каналы длинных костей для их стабилизации и укрепления. Его лечение оказалось чрезвычайно полезным в реабилитации и профилактике переломов; он был принят во всем мире и до сих пор является основой ортопедического лечения ОИ.
Спондилодез может быть выполнен для исправления сколиоза, хотя врожденная хрупкость костей делает эту операцию более сложной у пациентов с ОИ. Операция по удалению базилярных оттисков может быть выполнена, если давление на спинной мозг и ствол мозга вызывает неврологические проблемы.
Физиотерапия используется для мягкого укрепления мышц и улучшения моторики, минимизируя риск переломов. Это часто включает гидротерапию, легкие упражнения с сопротивлением и использование поддерживающих подушек для улучшения осанки. Людям рекомендуется регулярно менять положение в течение дня, чтобы сбалансировать задействованные мышцы и кости, находящиеся под давлением.
Обычно рекомендуются упражнения.
С адаптивным оборудованием, таким как костыли, инвалидные коляски с электроприводом, шины, захватные руки или модификации дома, многие люди с ОИ могут сохранять значительную степень автономии.
Более чем у 1 из 2 человек с ОИ также имеется несовершенный дентиногенез (DI) - врожденное нарушение образования дентина. Стоматологическое лечение может стать проблемой из-за различных деформаций скелета и зубов, вызванных ОИ. Детям с НО следует пройти осмотр у стоматолога, как только у них прорезываются зубы, это может минимизировать потерю структуры зубов в результате аномального дентина, и за ними следует регулярно наблюдать, чтобы сохранить их зубы и здоровье полости рта.
Многие люди с НО лечатся бисфосфонатами, и есть несколько осложнений стоматологических процедур, когда человек принимает АД, а именно: связанный с бисфосфонатом остеонекроз челюсти (BRONJ).
Состояние или его типы на протяжении многих лет и в разных странах носили различные названия. Среди наиболее распространенных альтернатив - синдром Экмана-Лобштейна, синдром Вролика и разговорная болезнь стеклянных костей. Название несовершенный остеогенез датируется по крайней мере 1895 годом и до сих пор является обычным медицинским термином в 20 веке. Текущая система четырех типов началась с Sillence в 1979 году. Более старая система считала менее тяжелые типы «поздним несовершенным остеогенезом», в то время как более тяжелые формы считались «несовершенным остеогенезом врожденным». Поскольку эта терминология плохо различается между типами, а все формы остеогенеза являются врожденными, это соглашение об именах с тех пор вышло из употребления.
Состояние было обнаружено в древнеегипетской мумии 1000 г. до н.э. норвежский король Ивар Бескостный, возможно, тоже имел это состояние. Самые ранние его исследования начались в 1788 г. у шведа Улофа Якоба Экмана. Он описал это состояние в своей докторской диссертации и упомянул случаи, когда оно восходит к 1678 году. В 1831 году описал это состояние на себе и двух братьях. Жан Лобштейн имел дело с этим у взрослых в 1833 году. Виллем Вролик действительно работал с этим заболеванием в 1850-х годах. Идея, что взрослая и новорожденная формы идентичны, возникла в 1897 году в работе Мартина Бенно Шмидта.
В Соединенных Штатах частота несовершенного остеогенеза оценивается в быть одним на 20 000 живорождений. По оценкам, от 20 000 до 50 000 человек страдают ОИ в Соединенных Штатах. Наиболее распространены типы I - IV, остальные очень редки.
Частота примерно одинакова для разных групп, но по неизвестным причинам шона и ндебеле из Зимбабве, похоже, имеют более высокое соотношение типа III к типу I, чем другие группы. Похожая картина была обнаружена в сегментах нигерийского и южноафриканского населения. В этих различных случаях общее количество ОИ всех четырех типов было примерно таким же, как и у любой другой этнической принадлежности.
Общество хрупких костей - благотворительная организация Великобритании, которая поддерживает людей с этим заболеванием.
Фонд OI Society of Australia был основан в 1977 году. Его цель - предлагать информацию о заболевании, поддерживать исследования и повышать осведомленность общественности о лицах с несовершенным остеогенезом.. Каждые два года фонд проводит конференцию для обсуждения образовательных мероприятий и поддержки Дня Вишбона.
Канадское общество несовершенного остеогенеза было основано в 1983 году, это международная некоммерческая организация, которая помогает с проживанием с детьми, страдающими ОИ. Они оказывают эмоциональную поддержку, поощряют и поддерживают канадские медицинские исследования причин ОИ для всех типов вовлеченных. Эта организация также хранит и обновляет библиотеку медицинских исследований и данных об этом заболевании для общественности.
У собак ОИ является аутосомно-рецессивным заболеванием, что означает, что будут затронуты собаки с двумя копиями аллеля. Бигли, стандартные жесткошерстные таксы, золотистые ретриверы, пудели, бедлингтон-терьеры, норвежские лосиные собаки, а также стандартная и миниатюрная гладкошерстная такса, как известно, являются возможными переносчиками ОИ, а также мыши и некоторые породы рыб. У золотистых ретриверов это вызвано мутацией COL1A1, а у гончих - COL1A2. Обнаружено, что отдельная мутация в гене SERPINH1 вызывает это состояние у такс. Многие племенные организации и ветеринары предлагают тесты на ОИ, чтобы определить, является ли собака переносчиком ОИ. Собак, гетерозиготных по ОИ, следует вязать только с не-носителями. Никогда не следует скрещивать гомозиготных носителей, за исключением случаев, когда они не являются носителями.
Животные модели НО имеют решающее значение для диагностики, лечения и возможного излечения их человеческих аналогов. Экспериментальные методы лечения и терапии, используемые на животных, играют важную роль в успешном лечении ОИ у людей. Хотя собаки, мыши, рыбы и люди не идентичны генетически, было официально признано, что некоторые модели животных представляют различные типы ОИ у людей. Исследования по лечению животных проводятся параллельно с успешным лечением ОИ у людей.
| Классификация | D |
|---|---|
| Внешние ресурсы |
| На Викискладе есть средства массовой информации, связанные с несовершенным остеогенезом. |