
Войти
| Миопатия Немалин | |
|---|---|
| Другие названия | Миопатия Немалин-стержень |
| Специальность | Неврология |
Немалиновая миопатия (также называемая стержневой миопатией или немалиновой стержневой миопатией ) - это врожденное нервно-мышечное заболевание со многими симптомами, которые могут проявляться, например, мышечной слабостью, гиповентиляция, нарушение глотания и нарушение речевой способности. Выраженность этих симптомов варьируется и может в некоторой степени меняться в течение жизни. Распространенность оценивается в 1 случай на 50 000 живорождений. Это наиболее распространенная недистрофическая миопатия.
«Миопатия» означает мышечное заболевание. Мышечные волокна человека с немалиновой миопатией содержат нитевидные стержни, иногда называемые немалиновыми телами. Хотя стержни являются диагностическим признаком заболевания, они, скорее, являются побочным продуктом процесса болезни, а не сами по себе вызывают какую-либо дисфункцию. Люди с немалиновой миопатией (НМ) обычно испытывают задержку двигательного развития или отсутствие двигательного развития в тяжелых случаях, и слабость может возникать во всех скелетных мышцах, таких как мышцы рук, ног, туловища, сгибателей шеи, горла и лица.. Слабость, как правило, более выражена в проксимальных мышцах, чем в дистальных. Глазные мышцы обычно сохраняются.
Клинически расстройство часто подразделяется на группы с широким диапазоном перекрывающейся степени тяжести, от самой тяжелой неонатальной формы, несовместимой с жизнью, до такой легкой формы, что ее невозможно диагностировать, поскольку человек, кажется, функционирует. на самом низком уровне нормальной силы и адекватности дыхания. Спорадическая немалиновая миопатия с поздним началом (SLONM) не является врожденным заболеванием и считается мышечной болезнью, отличной от NM, которая возникает при рождении или в раннем детстве. Респираторные проблемы обычно являются основной проблемой для людей со всеми формами ЯМ, и респираторные инфекции - довольно распространенное явление. NM сокращает продолжительность жизни, особенно в более тяжелых формах, но агрессивный и проактивный уход позволяет большинству людей выжить и даже вести активный образ жизни.
Немалиновая миопатия - одно из нервно-мышечных заболеваний, охватываемых Ассоциацией мышечной дистрофии в США.
Знаки различаются от человека к человеку. Маленькие дети и младенцы не двигаются, им трудно есть и дышать. У маленьких детей, которым не поставлен диагноз сразу при рождении, это обычно первые видимые симптомы. Один из признаков - опухшее лицо на непропорциональных участках. Другие примеры у новорожденных включают покачивание и затруднения при движении. Другие симптомы включают ослабление мышц шеи и верхней части грудной клетки. У взрослых наиболее частым симптомом являются проблемы с дыханием. Другие симптомы у взрослых могут варьироваться от легких до тяжелых нарушений речи. Часто сколиоз ставят по сравнению с немалиновой миопатией. Поскольку младенцы с ЯМ развиваются и достигают совершеннолетия, когда они должны начать ходить, многим требуется больше времени, чем обычно, из-за отсутствия мышц или просто мышечной усталости.
Поскольку лицевые мышцы участвуют в поглощении НМ, удлиненные лица и нижняя челюсть часто наблюдаются у людей с ЯМ. Люди, пораженные ЯМ, обычно начинают чувствовать мышечное истощение в возрасте от 20 до 50 лет. Поскольку НМ становится только хуже, даже после лечения люди, у которых проявляются ранние признаки НМ, только быстрее становятся слабее, чем, например, подросток, у которого только сейчас проявляются симптомы. Гастроэзофагеальный рефлюкс, хотя и не часто, но связан с ЯМ. Сердечные аномалии могут возникать в результате НМ, но вероятность этого невысока.
Большинство детей с легкой формой НМ в конечном итоге ходят самостоятельно, хотя часто в более позднем возрасте. чем их сверстники. Некоторые используют инвалидные коляски или другие приспособления, такие как ходунки или скобы, для повышения своей мобильности. Люди с тяжелой формой НМ обычно имеют ограниченные движения конечностей и постоянно пользуются инвалидными колясками. Из-за слабости в мышцах туловища люди с НМ склонны к сколиозу, который обычно развивается в детстве и ухудшается в период полового созревания. Многие люди с ЯМ подвергаются операции спондилодеза для выпрямления и стабилизации спины. Остеопороз также распространен при НМ.
Хотя пациенты с раннего возраста часто имеют подвижность суставов, выходящую за пределы нормы, с возрастом, деформации суставов и сколиоз обычно возникают. Если человек с немалиновой миопатией внимательно следит за своими суставами на ранней стадии, проблемы с ними могут быть обнаружены в самом начале, а их прогрессирование может быть отложено. Лечение проблем с суставами варьируется от упражнений на растяжку с физиотерапией до хирургического введения брекетов. Польза физических упражнений у людей с немалиновой миопатией все еще изучается, однако исследователи заметили улучшение мышечной функции в результате упражнений низкой интенсивности. Следует избегать энергичных упражнений и использования тяжелых весов.
Внимание к респираторным заболеваниям имеет решающее значение для здоровья всех людей с ЯМ. Младенцы с тяжелой формой НМ часто испытывают респираторный дистресс при рождении или вскоре после него. Многие из них вентилируются через трахеостомию, и при правильной помощи при дыхании они могут достичь хорошего здоровья. Хотя респираторный компромисс может не сразу проявляться у людей с промежуточным или легким НМ, он почти всегда существует в той или иной степени. Как и при многих нервно-мышечных расстройствах, гиповентиляция может начаться незаметно, и она может вызвать серьезные проблемы со здоровьем, если ее не исправить с помощью неинвазивных механических устройств для облегчения дыхания, особенно ночью.
Слабость бульбарных (глоточных) мышц является основным признаком немалиновой миопатии. Большинство людей с тяжелой формой НМ не могут глотать и получать питание через зонд для кормления. Большинство людей со средней и легкой степенью НМ частично или полностью принимают пищу орально. Нарушение бульбарных мышц также может привести к затруднениям в общении. Люди с НМ часто имеют гиперназальную речь в результате плохого закрытия небно-глоточного порта (между мягким небом и задней частью глотки). Коммуникативные навыки могут быть улучшены с помощью логопеда, оральных протезов, хирургии и дополнительных устройств общения. Люди с НМ обычно очень общительны и умны, с большим желанием общаться.
Физическое проявление немалиновой миопатии сильно различается, но слабость обычно сосредоточена в проксимальных мышцах, особенно в дыхательных, бульбарных мышцах и мышцах туловища. У людей с тяжелой ЯМ проявляются явные симптомы при рождении, в то время как у людей с средней или легкой степенью НМ могут изначально не проявляться симптомы. Младенцы с НМ часто бывают «гибкими» и гипотоническими. Дети, рожденные с НМ, часто набирают силу по мере роста, хотя влияние мышечной слабости на особенности тела со временем может стать более очевидным. Взрослые с НМ обычно имеют очень стройное телосложение.
Миопатия Немалин вызывается мутациями в одном из по меньшей мере 11 различных генов. Немалиновая миопатия - это клинически и генетически гетерогенное заболевание, при котором могут встречаться как аутосомно-доминантная, и аутосомно-рецессивная формы. Диагноз ставится на основании клинических признаков, таких как мышечная слабость, отсутствие или низкие глубокие сухожильные рефлексы (гипорефлексия) и высокое сводчатое небо вместе с электронно-плотными агрегатами, называемыми немалиновыми палочками, которые наблюдаются на микроскопическом уровне внутри мышечных волокон. Генетическое подтверждение посредством идентификации известной генетической мутации у пациента также является важным компонентом диагностики.
Две наиболее распространенные генные мутации, вызывающие миопатию немалина, обнаруживаются на NEB или ACTA1.. Мутации гена NEB обычно приводят к появлению симптомов, присутствующих при рождении или начинающихся в раннем детстве. Эта мутация приводит к примерно 50% пациентов с немалиновой миопатией. Наиболее распространенный путь наследования для людей с мутациями в NEB - аутосомно-рецессивный, при котором каждый родитель несет одну мутированную копию вместе с одной нормально функционирующей копией гена, и они передают мутированную копию своему потомству. В некоторых случаях, иногда при мутации ACTA1, NM может быть вызван типом наследования аутосомного доминирования. Эта мутация вызывает от 15 до 25 процентов случаев ЯМ. Одна из причин, почему это ниже, заключается в том, что NM связан с мутациями de novo в ACTA1, спонтанно возникающими в яйцеклетке или сперматозоиде. Когда заболевание передается по наследству, каждая беременность с одними и теми же партнерами имеет одинаковый риск передачи мутировавших генов потомству. Новые мутации (de novo) также могут возникать, вызывая NM, и de novo мутации чаще всего обнаруживаются в гене ACTA1. MYPN - последний обнаруженный ген, связанный с NM. Риск всех случаев немалиновой миопатии одинаков у мужчин и женщин.
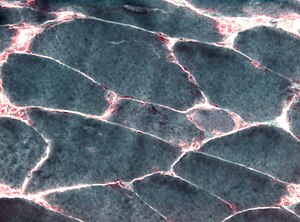 Пример мышечных клеток с палочками. Палочки окрашены в красный цвет и расположены на внутренней стороне плазматической мембраны.
Пример мышечных клеток с палочками. Палочки окрашены в красный цвет и расположены на внутренней стороне плазматической мембраны. | Ген 1 | Доля немалиновой миопатии, приписываемая мутации этого гена | Метод тестирования |
|---|---|---|
| NEB | До 50% | Анализ последовательности |
| Анализ деле / дупликации и целевой анализ для патогенных вариантов | ||
| ACTA1 | 15% - 25% | Анализ последовательности |
| Анализ делеции / дупликации | ||
| TPM3 | 2% -3% | Анализ последовательности |
| Анализ делеции / дупликации | ||
| TPM2 | <1% | Анализ последовательности |
| Анализ делеции / дупликации | ||
| TNNT1 | Практически исключительно у старых амишей | Анализ последовательности |
| Анализ деле / дупликации | ||
| CFL2 | Редко | Анализ последовательности |
| KBTBD13 | Неизвестно | Анализ последовательности |
| KLHL40 | 20% | Анализ последовательности |
| KLHL41 | Неизвестно | Анализ последовательности |
| LMOD3 | Неизвестно | Анализ последовательности |
| Неизвестно | NA | NA |
Физические возможности данного человека с ЯМ плохо коррелируют ни с генотипом, ни с мышечной патологией, наблюдаемой при биопсии.
Мышечные клетки сокращаются в сложных механических и химических процессах. Если какая-либо часть процесса или структуры нарушена, вероятно, возникнет дисфункция, как в случае с генетическими вариациями. У пациентов с немалиновой миопатией сокращаются мышцы. На электронно-микроскопическом уровне стержневидные компоненты часто можно увидеть в некоторых мышечных клетках, и если их увидеть, они являются диагностическими для состояния, называемого миопатией немалинового стержня. Наличие этих стержней само по себе не вызывает мышечной слабости; скорее они появляются в результате того, что в мышечном волокне что-то не так. Нет никакой связи между количеством стержней, обнаруженных в мышечных клетках, и степенью слабости человека. Все различные генные мутации, приводящие к состоянию, называемому немалиновой миопатией, которые были обнаружены до сих пор, находятся в генах, которые кодируют различные компоненты саркомера. В нормальных мышечных клетках различные части мышечных волокон, составляющих саркомер, равномерно распределены по схеме для эффективного сокращения мышц. Данные свидетельствуют о том, что некоторые виды ЯМ влияют на расположение этих мышечных волокон, в результате чего мышцы не могут сокращаться так же эффективно или эффективно.
 Это простой пример здорового саркомера.
Это простой пример здорового саркомера.  Пример саркомера с немалиновыми стержнями.
Пример саркомера с немалиновыми стержнями. Немалиновая миопатия обычно является генетической и проявляется у пораженного человека с рождения или с раннего возраста. Однако в некоторых случаях симптомы немалиновой миопатии не проявляются до зрелого возраста. Эти случаи обычно не являются генетическими. Из генов, связанных с немалиновой миопатией, большинство также участвует в кодировании белков саркомеров в мышечных клетках. Респираторные мышцы часто поражаются сильнее, чем другие группы скелетных мышц. При немалиновой миопатии сердечная мышца обычно не поражается; однако в тех случаях, когда это происходит, пациенты часто обращаются с дилатационной кардиомиопатией. Глазные мышцы обычно сохраняются.
Различные гены, мутации которых приводят к различным видам немалиновых миопатий, по-разному влияют на клетки и тело человека. Первый идентифицированный вид немалиновой миопатии связан с геном медленного α-тропомиозина TPM3 и варьируется от случая к случаю в зависимости от степени тяжести. При этом виде немалиновой миопатии пораженные люди слабее и сильнее поражаются нижними конечностями, чем верхними.
Как указано выше, наиболее распространенная генетическая форма НМ вызывается мутацией в гене небулина, называется Небулин, и имеет ряд уровней серьезности. Все опубликованные до этого момента случаи, когда НМ, как полагают, вызывается мутацией в гене NEB, были аутосомно-рецессивными и являются наиболее частой причиной немалиновой миопатии. У пациентов с этим типом ЯМ больше поражаются мышцы головы, чем проксимальные мышцы ядра тела. Следовательно, пациенты с этой генетической мутацией часто не могут поднять голову и говорить гнусавым голосом. Были случаи, когда предполагалось, что этот тип ЯМ может привести к более высокому интеллекту.
Третий вид немалиновой миопатии в скелетных мышцах ген α-актина ACTA1 возникает из-за рецессивного нулевая мутация. У этих пациентов не всегда наблюдаются типичные немалиновые тельца в мышечных клетках. Единственная аномалия, которую они показывают, - это неправильное распределение мышечных волокон.
Есть несколько других идентифицированных видов мутаций, которые приводят к миопатиям Немалина. Один влияет на медленные скелетные мышцы, другой приводит к образованию как немалиновых тел, так и других аномальных, стержневидных структур, образующихся в мышцах пациента.
В настоящее время миопатия Немалин не лечится. Немалиновая миопатия - очень редкое заболевание, которым в среднем страдает только 1 из 50 000, хотя недавние исследования показывают, что это число еще меньше. Существует ряд методов лечения, позволяющих свести к минимуму симптомы болезни. Лечение и процедуры, помогающие пациентам с немалиновой миопатией, различаются в зависимости от тяжести заболевания. Возможным приспособлением может быть использование стабилизатора, такого как скоба. Другие средства включают умеренную растяжку и умеренные упражнения, которые помогают целевым мышцам поддерживать максимальное здоровье. Поскольку люди с ЯМ растут и развиваются на протяжении всей своей жизни, для них важно регулярно посещать различных медицинских специалистов, включая невролога, физиотерапевта и других, например логопедов и психологов, чтобы помочь пациенту и его семье приспособиться к повседневной жизни.
Хотя нет лекарства от НМ, он Это возможно и является обычным явлением для многих людей, ведущих здоровый активный образ жизни даже в умеренных и тяжелых случаях. Исследования продолжают искать способы облегчить изнуряющие симптомы и качественно продлить продолжительность жизни пострадавших. Некоторые люди заметили легкие улучшения в обработке секреции, уровне энергии и физическом функционировании с добавлением L-тирозина, аминокислоты, которую можно приобрести в медицинских центрах. Некоторые симптомы могут ухудшаться с возрастом пациента. Потеря мышечной массы увеличивается с возрастом естественным образом, но еще более значительна при немалиновой миопатии.
Новые исследовательские ресурсы стали доступны сообществу NM, например CMDIR (реестр) и CMD-TR (биорепозиторий). Эти два ресурса объединяют семьи и отдельных лиц, заинтересованных в участии в исследованиях, с учеными, которые стремятся лечить или лечить ЯМ. Некоторые исследования НМ направлены на то, чтобы лучше понять молекулярные эффекты, которые мутации генов оказывают на мышечные клетки и остальное тело, и выявить любые связи, которые могут иметь НМ с другими заболеваниями и осложнениями со здоровьем.
«стержневидная миопатия» была впервые определена Дугласом Рей, австралийским врачом, в 1958 году. Однако результаты Рейя так и не были опубликованы, потому что другой врач отклонил его открытие. палочек в мышечной ткани как артефакт биопсии. Сорок лет спустя у пациента с «стержневой миопатией» Рея была подтверждена миопатия немалина. Другая группа австралийских исследователей с тех пор опубликовала статью, в которой признает Рейя за его работу.
«Немалиновая миопатия» впервые была названа в опубликованной в 1963 году статье североамериканскими исследователями и Дж. М. Шай. Шай и его команда обнаружили стержневидные структуры в мышечных волокнах пациентов с мышечной слабостью, выполнив биопсию мышц у нескольких пациентов. Лаборатории, проводящие исследования ЯМ, расположены по всему миру, особенно в США, Канаде, Англии, Финляндии и Австралии.
В 1999 году был запущен первый веб-сайт, посвященный немалиновой миопатии, а в октябре 2004 года в Торонто, Канада, была проведена первая конференция по немалиновой миопатии. С тех пор было проведено еще много конференций и общественных мероприятий, и все мероприятия, организованные с 2008 года, спонсировались Фондом укрепления силы немалиновой миопатии (AFBS), единственным фондом, ориентированным на поддержку разработки методов лечения и социальных мероприятий для сообщества NM. В марте 2006 года Ники Шислер выпустила книгу «Хрупкое», в которой она рассказала о своем опыте рождения сыновей-близнецов с тяжелой формой НМ. В 2014 году группа экспертов в сотрудничестве с больными людьми и семьями, ухаживающими за людьми с врожденной миопатией, разработала первое руководство по управлению жизнью с врожденной миопатией
| Классификация | D |
|---|---|
| Внешние ресурсы |