
Войти
| Медуллярная кистозная болезнь почек | |
|---|---|
 | |
| Медуллярная кистозная болезнь почек имеет аутосомно-доминантный тип наследования. | |
| Специальность | Медицинская генетика |
| Симптомы | Полидипсия |
| Типы | MCKD1 и MCKD2 |
| Диагностический метод | Биопсия почек, УЗИ почек, Общий анализ крови |
| Медикамент | В настоящее время нет лекарства, Пейте много жидкости, Солевые добавки |
Медуллярная кистозная болезнь почек (MCKD) - аутосомно-доминантное заболевание почек, характеризующееся тубулоинтерстициальным склерозом, приводящим к терминальной стадии почечной недостаточности. Поскольку наличие кист не является ни ранним, ни типичным диагностическим признаком заболевания, и поскольку по крайней мере 4 различных генных мутации могут вызывать это состояние, было предложено добавить название аутосомно-доминантная тубулоинтерстициальная болезнь почек (ADTKD). с лежащим в основе генетическим вариантом для конкретного человека. Важно отметить, что если кисты обнаруживаются в мозговых собирательных протоках, они могут привести к сморщенной почке, в отличие от поликистоза почек. Существуют две известные формы медуллярной кистозной болезни почек: болезнь почек, вызываемая муцином-1 (MKD1), и болезнь почек, вызванная муцином-2 / уромодулиновая болезнь почек (MKD2). Третья форма заболевания возникает из-за мутаций в гене, кодирующем ренин (ADTKD-REN), и ранее была известна как семейная ювенильная гиперурикемическая нефропатия 2 типа.
Что касается признаков / симптомов медуллярной кистозной болезни почек, это заболевание нелегко диагностировать и встречается редко. В этом состоянии потеря функции почек происходит медленно с течением времени, однако у пораженного человека могут наблюдаться следующие признаки / симптомы:
У некоторых людей с этим заболеванием развивается подагра - состояние, при котором у пациентов развиваются сильная боль и отек большого пальца ноги или другого сустава, такого как колено. При отсутствии лечения он становится хроническим и чаще всего поражает суставы, а не периодически.
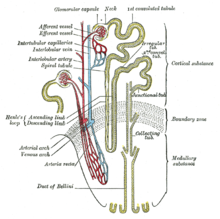 Проксимальный извитый каналец
Проксимальный извитый каналец Нормальные люди имеют две копии генов MUC1 и UMOD. Гены продуцируют белок муцин-1 и уромодулин соответственно. Эти белки экспрессируются только в определенных клетках почек - толстой восходящей конечности Генле и дистальном извитом канальце - обеих частях почечного канальца. Белок покрывает поверхность канальца и защищает канальец.
При МКБ у людей есть один нормальный и один аномальный ген MUC1. При уромодулиновой болезни почек (UKD) люди имеют один нормальный и один аномальный ген UMOD. Аномальный (мутировавший) ген производит неправильно свернутый белковый продукт, который откладывается в клетках почечных канальцев (в части клетки, называемой эндоплазматическим ретикулумом). Аномальный белок накапливается в клетке и заставляет ее медленно умирать, что приводит к окончательной потере функции почек, что проявляется в болезненном состоянии.
 1. Клубочки, 2. Эфферентная артериола, 3. Капсула Боумена, 4. Проксимальный извитый каналец, 5. Кортикальный собирательный канал, 6. Дистальный извитый каналец, 7. Петля Генле, 8. Папиллярный проток, 9. Перитубулярные капилляры, 10. Дугообразная вена, 11. Дуговидная артерия, 12. Приводящая артериола, 13. Юкстагломерулярный аппарат.
1. Клубочки, 2. Эфферентная артериола, 3. Капсула Боумена, 4. Проксимальный извитый каналец, 5. Кортикальный собирательный канал, 6. Дистальный извитый каналец, 7. Петля Генле, 8. Папиллярный проток, 9. Перитубулярные капилляры, 10. Дугообразная вена, 11. Дуговидная артерия, 12. Приводящая артериола, 13. Юкстагломерулярный аппарат. Заболевание почек Mucin-1 (MKD) возникает из-за мутации в гене MUC1, который расположен на хромосоме 1. Белок муцин-1 участвует в создании слизеподобного вещества, которое покрывает поверхность различных мелких канальцев в организме, он экспрессируется на клетках дистальных канальцев в почках. Заболевание почек, вызванное муцином-1 (MKD), вызывается мутацией в гене MUC1.
Эта мутация изменяет генетическую последовательность в гене MUC1, что приводит к появлению нового мутировавшего белка. Кроме того, заболевание встречается крайне редко, и его распространенность неизвестна. Это заболевание является аутосомно-доминантным, что означает, что для него характерна вероятность наследования 50% и медленно прогрессирующее хроническое заболевание почек, которое приводит к необходимости диализа или трансплантации почки. Люди с мутацией в этом гене могут иметь переменную скорость потери функции почек: некоторые люди идут на диализ в возрасте от тридцати лет, в то время как другие могут не идти на диализ до более позднего возраста.
Ранее это состояние называлось медуллярной кистозной болезнью почек типа 1. Оно было названо так, потому что у некоторых людей с этим заболеванием были кисты (маленькие отверстия) в середине (мозговом веществе) почек. С тех пор было обнаружено, что эти кисты встречаются редко и не обнаруживаются у большинства пациентов с мутациями MUC1. По этой причине от этого названия отказались, ученые (входящие в группу «Инициативы по диализу почек и глобальные результаты»), специализирующиеся на этом заболевании, официально объединились и создали официальное название для этого и подобных состояний.
Это состояние было обозначено как аутосомно-доминантное тубулоинтерстициальное заболевание почек (ADTKD). При аутосомно-доминантном наследовании необходимо затронуть только одного родителя, чтобы передать заболевание. Тубулоинтерстициальный препарат используется потому, что проблемы, возникающие при этом заболевании, являются следствием атрофии канальцев, петли Генле и фиброза медуллярного интерстиция.
 Хромосома 16
Хромосома 16 Медуллярная кистозной почек болезни типа 2 происходит из - за мутаций в гене под названием UMOD на хромосоме 16, который кодирует белок, называемый uromodulin Это заболевание также аутосомно - доминантный, а это означает, что она характеризуется 50% вероятность наследования и медленно прогрессирующим хроническим заболеванием почек этой приводит к необходимости диализа или пересадки почки. Люди с этим заболеванием относительно рано болеют подагрой. Мутации в этом гене могут приводить к различной степени потери функции почек.
При наследовании MKD2, как и MKD1, аутосомно-доминантный характер указывает на то, что 50% детей пострадавшего человека унаследуют болезнь. «Тубулоинтерстициальный» используется потому, что проблемы, возникающие при этом заболевании, возникают в отделе почек, известном как тубулоинтерстиций, у большинства людей используется более короткое название заболевания - уромодулин почечная болезнь (UKD).
 Биопсия почки / микрофотография
Биопсия почки / микрофотография Диагноз медуллярной кистозной болезни почек может быть поставлен с помощью медицинского осмотра. Дальнейшие тесты / экзамены заключаются в следующем:
Что касается лечения / ведения медуллярной кистозной болезни почек, в настоящее время нет никаких специфических методов лечения этого заболевания, и нет никаких известных диет, замедляющих прогрессирование заболевания. Однако лечение симптомов может осуществляться следующим образом: эритропоэтин используется для лечения анемии, а гормон роста используется, когда рост становится проблемой. Кроме того, в какой-то момент может потребоваться пересадка почки. Наконец, необходимо сократить потребление продуктов, содержащих калий и фосфаты.
Ученые из Института Броуда в Кембридже, штат Массачусетс, определили генетическую причину UKD как мутации в гене MUC1.
| Классификация | D |
|---|---|
| Внешние ресурсы |
| У Схолии есть тематический профиль медуллярной кистозной болезни почек. |